CellCharter 分析
前言
IMPORTANT
CellCharter 是一种专门用于空间转录组学数据分析的工具,通过邻域富集分析揭示细胞在组织中的空间相互作用模式。它能够识别细胞类型之间的空间共定位关系,为理解组织微环境和细胞间通讯提供重要线索。
在空间转录组学研究中,我们不仅关注基因表达模式,更希望理解细胞在空间上的分布规律和相互作用。CellCharter 通过构建空间网络并计算邻域富集分数,能够定量分析细胞类型间的空间邻近性,为解析组织结构和功能提供重要工具。
CellCharter 的核心功能
- 空间网络构建:基于细胞或斑点的空间坐标构建空间邻近网络
- 邻域富集分析:计算细胞类型间的空间共定位程度
- 差异空间分析:比较不同条件下细胞空间分布的变化
- 可视化展示:提供直观的热图展示空间相互作用模式
本篇文档旨在为空间转录组学研究者提供一份详尽的 CellCharter 技术指南,内容涵盖其基本原理、在 SeekSoul Online 云平台上的操作方法、结果解读、实战案例及常见问题,帮助您快速掌握并应用该工具。
CellCharter 理论基础
核心原理
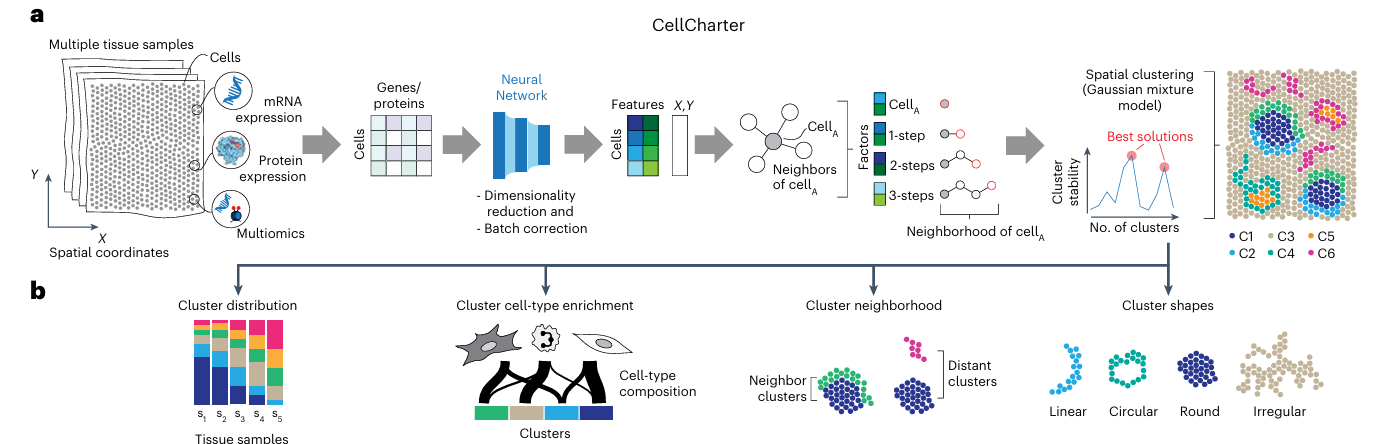
CellCharter 的核心思想是:通过构建空间网络并计算邻域富集分数,定量分析细胞类型间的空间共定位关系。这一过程可以概括为以下几个主要步骤:
- 空间网络构建:根据细胞或斑点的空间坐标构建空间邻近网络
- 邻域富集计算:计算两个细胞群之间的实际连接数与随机预期的比值
- 富集分数评估:通过邻域富集分数 (NE) 评估空间共定位程度
- 差异分析:比较不同条件下的空间相互作用变化
关键算法详解
空间网络构建
- 原理:基于细胞或斑点的空间坐标,使用 Delaunay 三角剖分构建空间邻近网络
- 方法:将每个细胞或斑点视为网络节点,根据空间距离确定节点间的连接
- 优势:能够准确反映细胞在组织中的真实空间关系
邻域富集分析
- 观察值 (Observed):两个细胞群之间的实际连接数
- 预期值 (Expected):基于节点度数的随机预期连接数
- 邻域富集分数:NE = Observed/Expected
- 解释:
- NE > 1:表示细胞群间空间富集(共定位)
- NE < 1:表示细胞群间空间排斥(分离)
- NE = 1:表示细胞群间空间随机分布
差异邻域富集分析
- 原理:比较不同条件下(如健康与疾病)的邻域富集分析结果
- 方法:计算差异邻域富集分数 (Differential NE)
- 统计检验:通过随机采样条件标签计算 P 值评估差异显著性
生物学意义
组织微环境解析
- 功能:识别组织中的功能区域和细胞间相互作用
- 应用:理解组织发育、疾病进展等生物学过程
细胞通讯研究
- 功能:揭示细胞间通过空间邻近性进行的通讯机制
- 应用:研究细胞信号传导和调控网络
云平台操作指南
在云平台上,CellCharter 分析流程被设计得直观易用。您无需编写代码,只需通过参数配置界面即可完成分析。
分析前的准备
IMPORTANT
CellCharter 分析的成功与否,很大程度上取决于输入数据的质量和空间信息的准确性。在开始分析前,请务必确认:
- 数据已完成预处理:您的空间转录组数据已经过标准的质控、降维、聚类和细胞类型注释。
- 空间坐标信息完整:确保每个细胞或斑点都有准确的空间坐标信息。
- 细胞类型注释准确:细胞类型注释的准确性直接影响邻域富集分析的结果。
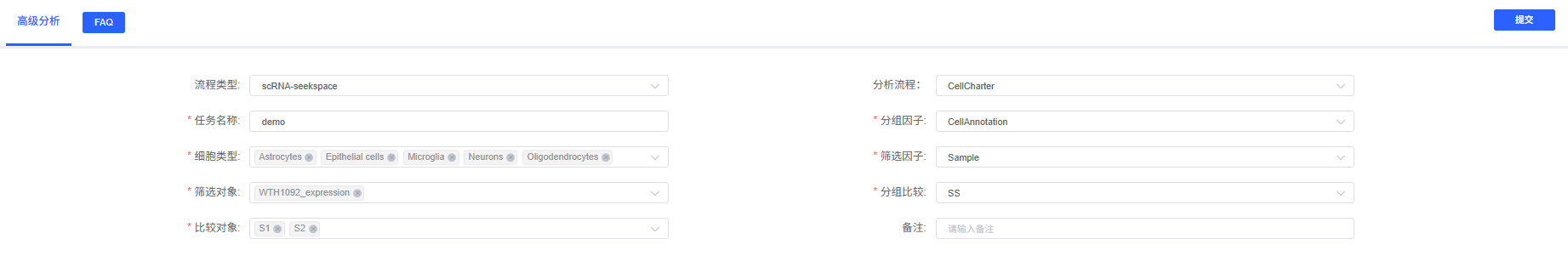
参数详解
下表详细列出了云平台 CellCharter 分析模块的主要参数及其说明。
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头,可包含英文字母、数字、下划线和中文。 |
| 分组因子 | meta 的列名,例如 CellAnnotation,必填。 |
| 细胞类型 | 基于 meta 的 col_celltype 列对应的对象,必填。 |
| 筛选因子 | meta 的列名,例如 Sample,必填。 |
| 筛选对象 | 基于 meta 的 col_sam 列所对应的对象,必填。 |
| 分组比较 | meta 的列名,例如 group,必填。 |
| 比较对象 | 基于 meta 的 col_group 列所对应的对象,必填。 |
| 备注 | 自定义备注信息。 |
重要注意事项
CAUTION
- 空间坐标要求:确保空间坐标信息准确且完整,缺失或错误的坐标信息会导致分析失败。
- 细胞类型选择:选择具有生物学意义的细胞类型进行分析,避免选择细胞数量过少的类型。
- 样本质量控制:确保样本质量良好,低质量样本会影响空间网络构建的准确性。
操作流程
- 进入分析模块:在云平台导航至"高级分析"模块,选择"CellCharter"。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息等。
- 提交任务:确认参数无误后,点击"提交"按钮,等待分析完成。
- 下载与查看:分析结束后,在任务列表中下载并查看生成的分析报告和结果文件。
结果解读
CellCharter 的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
结果文件列表
| 文件名 | 内容说明 |
|---|---|
*_enrichment_heatmap.png | 细胞类型邻域富集热图(PNG 格式) |
*_enrichment_heatmap.pdf | 细胞类型邻域富集热图(PDF 格式) |
*_enrichment_heatmap.txt | 邻域富集分数数据表格 |
邻域富集热图解读
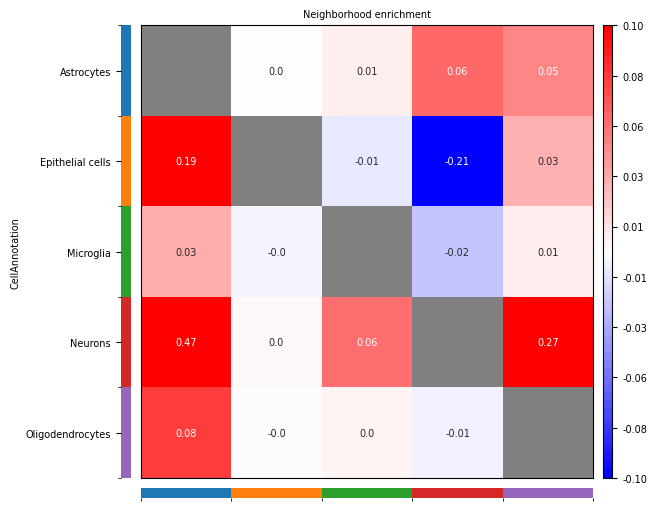
热图结构
- 行:源细胞类型 (Ci)
- 列:目标细胞类型 (Cj)
- 数值:邻域富集分数 (NE)
颜色含义
- 正数(红色):表示细胞群 Ci 与细胞群 Cj 在空间上倾向于富集,即更有可能相邻
- 负数(蓝色):表示细胞群 Ci 与细胞群 Cj 在空间上倾向于排斥,即更有可能分离
- 零(白色):表示细胞群 Ci 与细胞群 Cj 的连接数与随机预期一致,没有明显的富集或排斥
生物学解释
- 空间富集:提示两种细胞类型可能存在功能相关性,如细胞间通讯、协同作用等
- 空间排斥:提示两种细胞类型可能存在功能拮抗或竞争关系
- 随机分布:提示两种细胞类型在空间上相互独立
数据表格解读
邻域富集分数数据表格包含以下信息:
- 行名:源细胞类型
- 列名:目标细胞类型
- 数值:对应的邻域富集分数
应用案例
案例一:CellCharter 方法性能比较分析
- 文献:Varrone, M., Tavernari, D., Santamaria-Martínez, A. et al. Nature Genetics. 2024.
- 背景:研究者使用人类背外侧前额叶皮层 (DLPFC) Visium 数据集,比较了 CellCharter 与其他空间聚类方法的性能。
- 分析策略:使用调整兰德指数 (ARI) 评估不同方法在空间聚类任务中的表现,包括 DR.SC、SOTIP、SEDR、BayesSpace、STAGATE、UTAG 等方法。
- 核心发现:
- CellCharter 在所有比较方法中表现最佳,平均 ARI 达到 0.62,显著优于其他方法。
- 在 GPU 和 CPU 环境下,CellCharter 都展现出优异的性能表现。
- 通过统计检验验证了 CellCharter 相对于其他方法的显著优势。
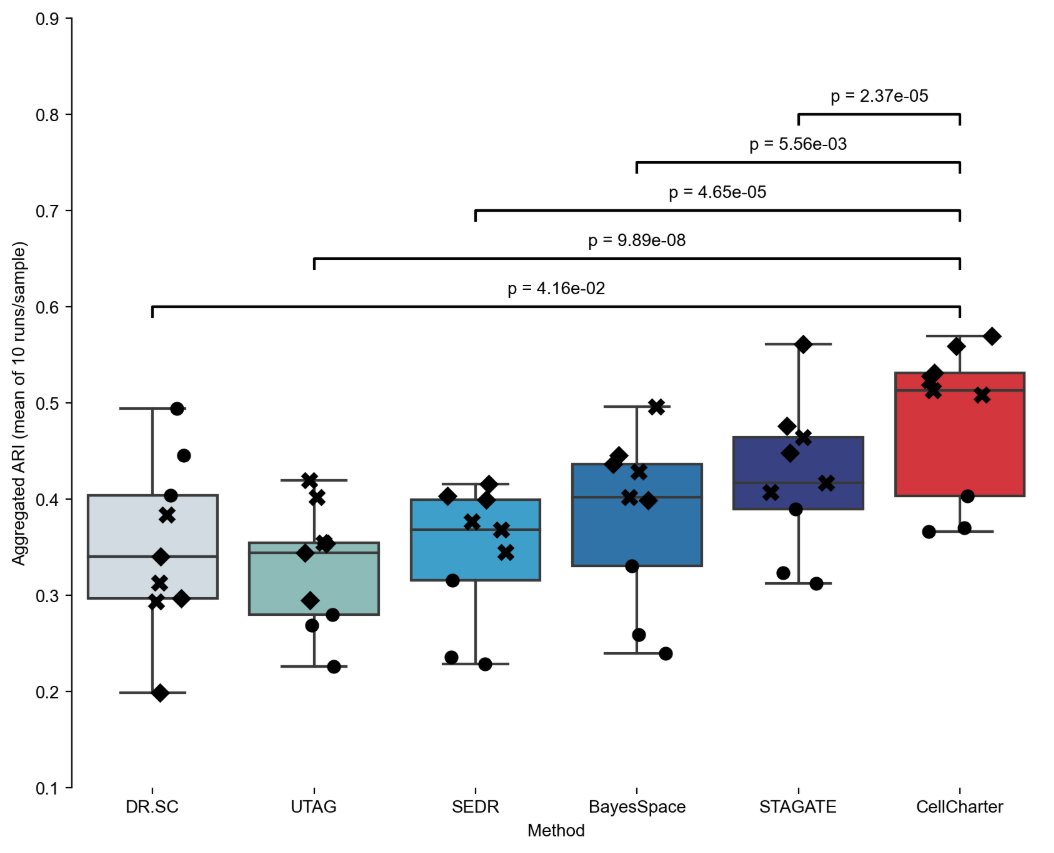
图:CellCharter 与其他空间聚类方法的性能比较。箱线图显示 CellCharter 在所有方法中表现最佳,平均 ARI 显著高于其他方法。图中还显示了不同患者样本的个体表现(用不同标记表示)。
案例二:人类大脑皮层的空间聚类分析
- 文献:Varrone, M., Tavernari, D., Santamaria-Martínez, A. et al. Nature Genetics. 2024. (Fig. 1)
- 背景:使用 CellCharter 分析人类背外侧前额叶皮层 (DLPFC) Visium 数据集,比较 CellCharter 与其他空间聚类方法的性能表现。
- 分析策略:对 DLPFC 样本进行空间聚类分析,使用调整兰德指数 (ARI) 评估不同方法的表现,包括 DR.SC、SOTIP、SEDR、BayesSpace、STAGATE、UTAG 等方法。
- 核心发现:
- CellCharter 在所有比较方法中表现最佳,平均 ARI 达到 0.62,显著优于其他方法。
- 相比其他方法,CellCharter 在保持空间连续性的同时,更好地捕获了皮层层的边界。
- 通过批次效应校正,CellCharter 在不同样本间保持了一致的聚类结果。
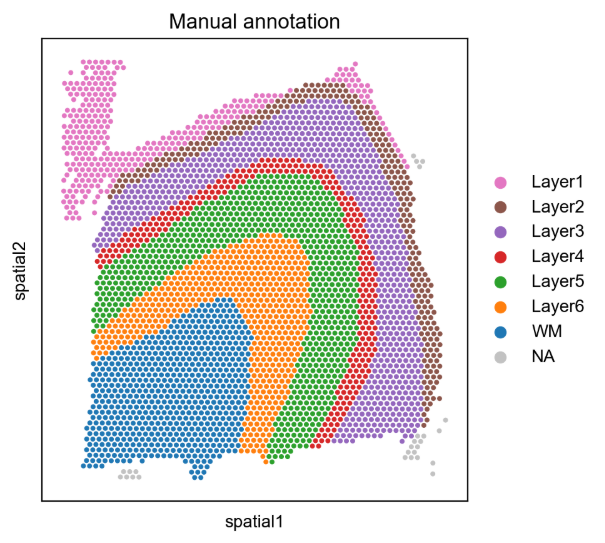
图:人类背外侧前额叶皮层的空间聚类结果比较。该图展示了 CellCharter 在 Visium 数据上的空间聚类性能,能够准确识别不同皮层层的空间分布模式。
案例三:小鼠脾脏 CODEX 数据的空间生态位分析
- 文献:Varrone, M., Tavernari, D., Santamaria-Martínez, A. et al. Nature Genetics. 2024. (Figs. 2-3)
- 背景:使用 CellCharter 分析小鼠脾脏 CODEX 数据中不同细胞类型的空间分布和相互作用模式,识别空间生态位结构。
- 分析策略:对 CODEX 数据进行空间聚类,计算细胞类型富集和邻域富集分析,比较 CellCharter 与其他方法的空间聚类性能。
- 核心发现:
- CellCharter 识别出 11 个不同的空间生态位,包括 B 细胞滤泡、PALS 区域、边缘区等。
- 相比 STAGATE 等其他方法,CellCharter 能够更清晰地识别出不同的空间生态位结构。
- 通过邻域富集分析揭示了不同细胞类型间的空间相互作用模式。
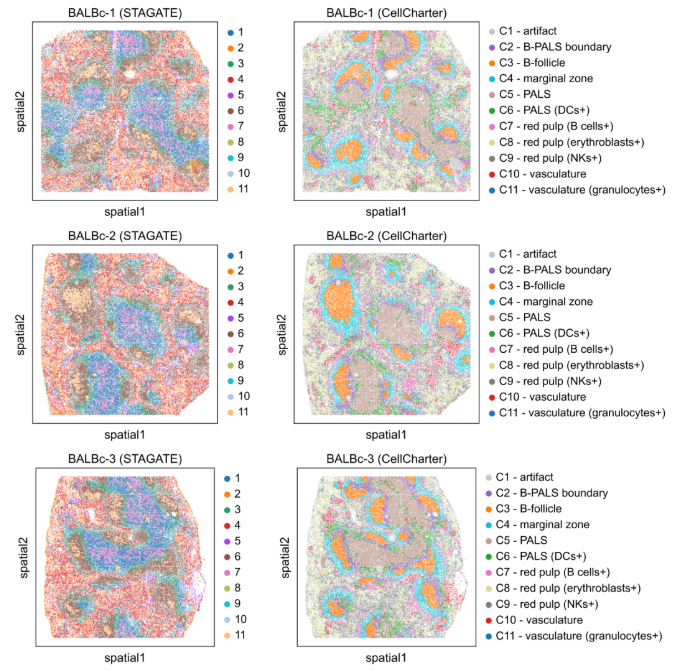
图:小鼠脾脏 CODEX 数据的空间聚类结果比较。上图显示 STAGATE 方法的结果,下图显示 CellCharter 的结果。CellCharter 能够更清晰地识别出不同的空间生态位,包括 B 细胞滤泡、PALS 区域等。
案例四:细胞类型邻域富集分析
- 文献:Varrone, M., Tavernari, D., Santamaria-Martínez, A. et al. Nature Genetics. 2024. (基于 CODEX 小鼠脾脏数据)
- 背景:使用 CellCharter 分析小鼠脾脏 CODEX 数据中不同细胞类型间的空间富集和排斥模式,揭示组织中的细胞-细胞相互作用。
- 分析策略:计算细胞类型间的邻域富集分数,生成富集热图,比较不同条件下的空间相互作用差异。
- 核心发现:
- 发现 B 细胞与 T 细胞在 PALS 区域存在显著的空间富集。
- 识别出不同免疫细胞亚群在脾脏中的特异性空间分布模式。
- 通过差异邻域富集分析揭示了疾病状态下的空间组织改变。
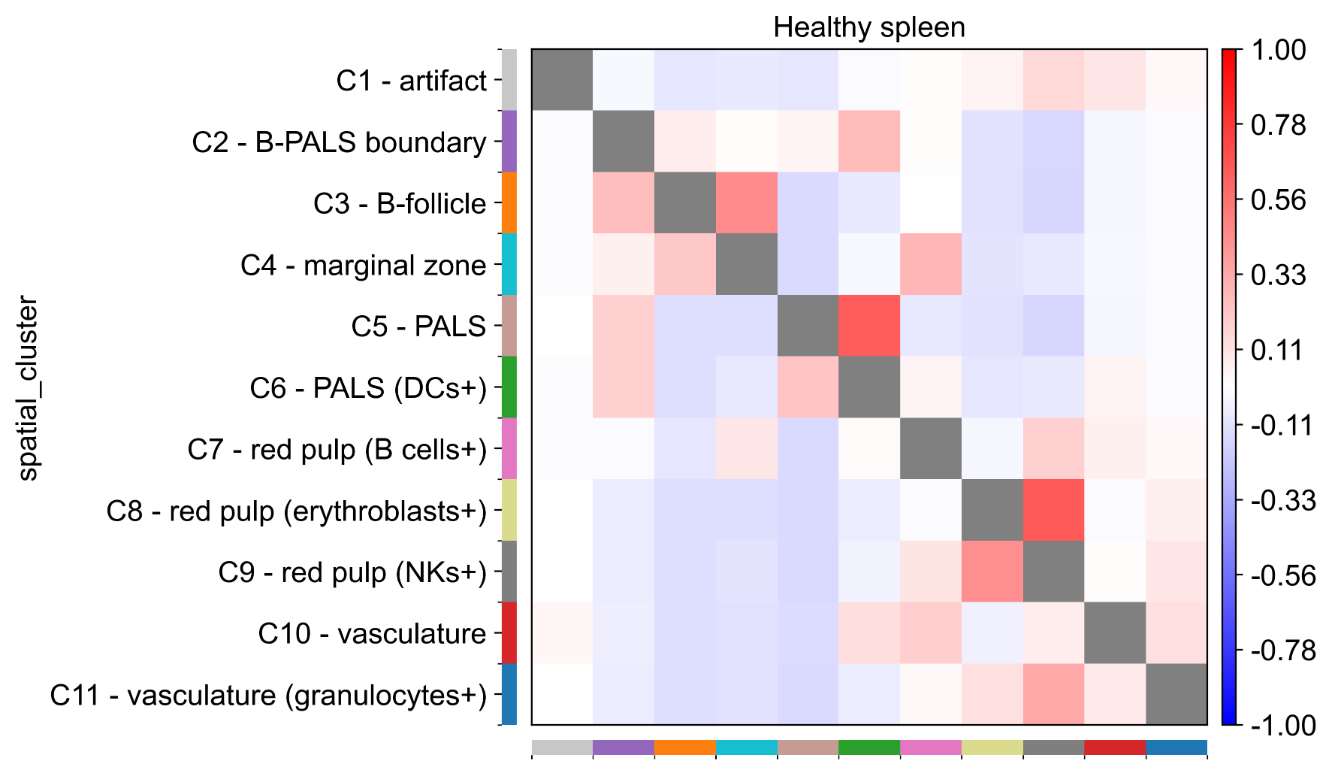
图:细胞类型邻域富集分析热图。红色表示空间富集,蓝色表示空间排斥。可以看出不同细胞类型间存在复杂的空间相互作用模式,反映了脾脏组织的空间组织结构。
注意事项与最佳实践
WARNING
避免过度解读:CellCharter 结果是基于空间邻近性的计算推断,不等于真实的生物学相互作用。任何关键发现都需要后续的生物学实验来证实。
CAUTION
数据质量至关重要:CellCharter 分析对空间坐标信息的准确性要求很高,低质量的空间数据可能导致假阳性结果。务必确保空间坐标信息准确且完整。
TIP
关注生物学意义:除了统计学显著性,更应关注空间相互作用的生物学功能和疾病相关性,结合功能富集分析和已知的细胞相互作用知识进行综合判断。
NOTE
结果受参数影响:CellCharter 分析结果会受到细胞类型注释的准确性以及分析参数(如选择的细胞群、空间网络构建方法)的影响。如果初步结果不理想,可以尝试调整输入参数重新进行分析。
常见问题解答 (FAQ)
Q1: CellCharter 分析需要多长时间?
A: 分析时间取决于数据规模和计算资源配置。一般来说:
- 小数据集 (1,000-5,000 细胞):30 分钟-1 小时
- 中等数据集 (5,000-20,000 细胞):1-3 小时
- 大数据集 (>20,000 细胞):3-8 小时或更长
Q2: 邻域富集分数的意义是什么?
A:
- NE > 1:表示两种细胞类型在空间上富集,存在共定位关系
- NE < 1:表示两种细胞类型在空间上排斥,相互分离
- NE = 1:表示两种细胞类型在空间上随机分布,无明显的空间关联
Q3: 如何判断空间相互作用的生物学意义?
A: 可通过以下方法判断空间相互作用的生物学意义:
- 功能富集分析:分析空间富集的细胞类型是否在功能上相关
- 文献验证:结合已知的细胞相互作用知识进行验证
- 差异分析:比较不同条件下空间相互作用的变化
Q4: 如何验证 CellCharter 分析结果的可靠性?
A: 可通过以下方式验证结果可靠性:
- 生物学验证:结合已知文献和数据库验证关键的空间相互作用
- 实验验证:通过免疫荧光、原位杂交等实验验证空间共定位
- 交叉验证:使用不同的数据集或分析方法验证结果一致性
参考资料
[1] VARRONE M, TAVERNARI D, SANTAMARIA-MARTÍNEZ A, et al. CellCharter reveals spatial cell niches associated with tissue remodeling and cell plasticity[J]. Nat Genet, 2024, 56: 74–84.
[2] CLARK D J, DATAR R, HISEY E, et al. Integrated proteogenomic characterization of clear cell renal cell carcinoma[J]. Cell, 2020, 180: 207-228.
[3] PALLA G, SPITZER H, KLEIN M, et al. Squidpy: a scalable framework for spatial omics analysis[J]. Nat Methods, 2022, 19: 171–178.
[4] WOLF F A, ANGERER P, THEIS F J. SCANPY: large-scale single-cell gene expression data analysis[J]. Genome Biol, 2018, 19: 15.
[5] PALLA G, FISCHER D S, LANGEN M, et al. Squidpy: a scalable framework for spatial omics analysis[J]. Nat Methods, 2022, 19: 171–178.