单细胞 scATAC-seq & scRNA-seq 双组学标准分析:GeneActivity 分析
前言
TIP
GeneActivity 分析通过计算基因体及其上游调控区域的染色质可及性,推断潜在的转录活性,以弥补单细胞 ATAC 数据中"只有开放区域,没有基因表达量"的信息缺口。
在单细胞多组学研究中,scATAC-seq 数据提供了染色质开放状态的信息,但无法直接反映基因的转录活性。GeneActivity(基因活力)分析通过统计每个基因的启动子(通常取 TSS 上游 2 kb)与基因体区域内的 fragment 数量,经过标准化后得到活性值,从而推断该基因是否具备转录潜力。
GeneActivity 的核心功能
基因活力推断
依据染色质可及性(ATAC-seq)数据,计算每个基因的潜在转录活性,为基因功能解读提供基础。基因活力差异分析
识别不同细胞群体间基因活力的差异。分析方法类似于差异表达(DEG)分析,通过比较不同细胞群体(如细胞类型、状态、处理组等)的基因活力值,筛选出在特定群体中显著高或低活力的调控基因。基因转录与基因活力相似性分析
对比不同细胞群在基因转录表达(scRNA-seq)与基因活力(scATAC-seq)层面的相关性,评估表观调控与基因表达间的一致性和差异性。
本篇文档旨在为单细胞多组学研究者提供一份详尽的 GeneActivity 技术指南,内容涵盖其基本原理、在 SeekSoulOnline 云平台上的操作方法、结果解读及常见问题,帮助您快速掌握并应用该工具。
GeneActivity 理论基础
核心原理
GeneActivity 分析的核心思想是:通过统计基因体及其上游调控区域内的染色质可及性信号(fragment 数量),推断该基因的潜在转录活性。 这个过程可以概括为三个主要步骤:
定义基因区域:确定每个基因的统计范围,通常包括基因体(gene body)和上游启动子区域(默认 TSS 上游 2 kb)。该范围既包含核心启动子也记录转录延伸阶段的可及性。
统计 fragment 数量:统计每个基因区域内每个细胞中的 fragment 数量。fragment 是 Tn5 转座酶在开放染色质区域切割所得的 DNA 片段,是 ATAC 信号的载体。
标准化输出:对统计得到的 fragment 计数进行标准化处理,生成基因活力矩阵。该矩阵的行对应基因,列对应单细胞,矩阵元素为基因区域的标准化 fragment 计数。
关键概念
基因活力 vs. 基因表达
| 指标 | 数据来源 | 反映信息 | 常见用途 |
|---|---|---|---|
| Gene Activity | scATAC-seq | 染色质开放程度 → 潜在转录能力 | 细胞类型注释、差异可及性基因、跨组学比对 |
| Gene Expression | scRNA-seq | mRNA 实际丰度 | 传统差异表达、功能富集、轨迹分析 |
TIP
GeneActivity 与 Gene Expression 通常正相关,但也会因表观遗传沉默、转录后调控或技术噪音而出现不一致。对比两者可更深入理解调控机制。
基因活力值的表示
GeneActivity 输出为稀疏矩阵,行对应基因、列对应单细胞,矩阵元素为基因区域的 fragment 计数。例如:
6 x 3 sparse Matrix of class "dgCMatrix"
AAACCCAATGCGTGGCA_1 AAACCCAATGTGCTACT_1 AAACCCACGACATCCGG_1
Hnf4g . . .
Zfhx4 . 5 2
Pex2 . . 2
UBC . . .
1700008P02Rik . . .
Pkia . . .调控区域覆盖
默认采用"基因体 + 上游 2 kb"框定统计范围,该范围兼顾启动子与转录延伸阶段,既具生物学意义又能缓解 ATAC 数据稀疏问题。上游扩展距离可根据物种或研究对象调整(1–3 kb 最常见),但需保持全数据分析的一致性。
差异 GeneActivity 分析
GeneActivity assay 构建完成后,可直接进行差异分析,识别不同细胞群体间基因活力的差异。差异分析的方法与 RNA 数据的差异基因分析方法类似,通过比较不同细胞群体(如不同细胞类型、不同处理组)的基因活力值,找到在特定群体中显著高活力的基因。
- 适用场景:一次性比较所有细胞群体,或指定两组完成疾病/处理 vs 对照的对比。
- 推荐筛选标准:
p_val_adj < 0.05、avg_log2FC > 0.25、pct_in > pct_out。
云平台操作指南
在云平台上,GeneActivity 分析流程被设计得直观易用。您无需编写代码,只需通过参数配置界面即可完成分析。
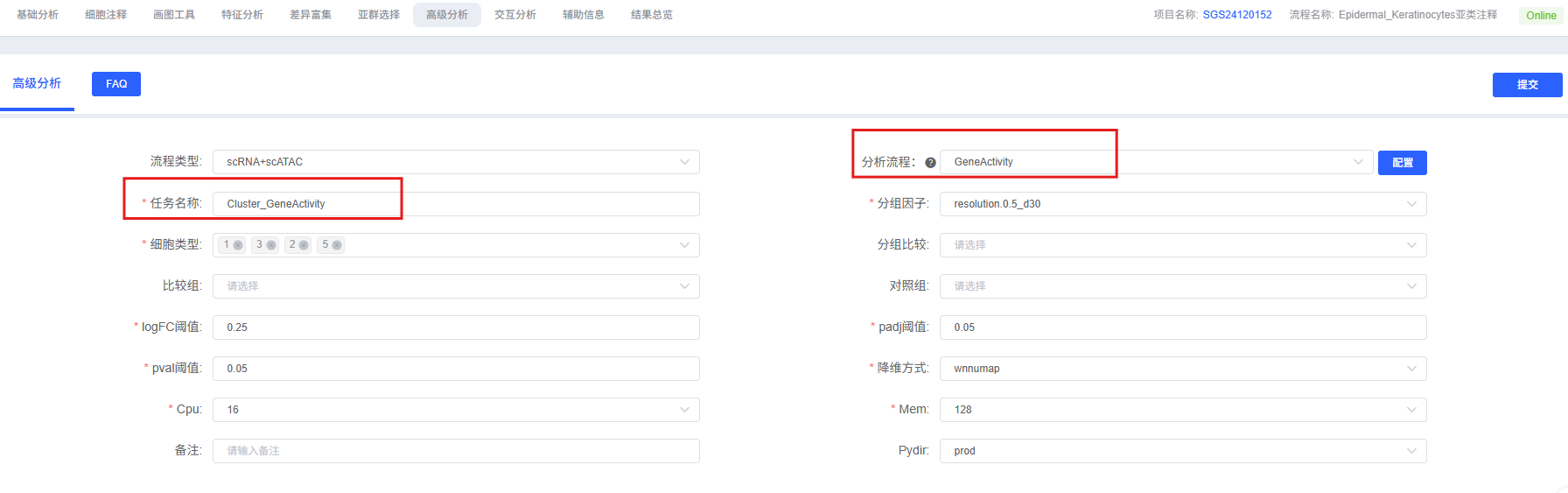
分析前的准备
TIP
GeneActivity 分析的成功与否,很大程度上取决于输入数据的质量和多组学数据的匹配度。在开始分析前,请务必确认:
- 数据已完成预处理:您的单细胞 ATAC 和 RNA 数据已经过标准的质控、降维、聚类和细胞类型注释。
- 数据匹配性:确保 ATAC 和 RNA 数据来自相同的细胞或匹配的细胞群体,以便进行跨组学比对。
- 注释信息完整:确保数据中包含必要的细胞类型、样本分组等注释信息,以便进行差异分析和结果展示。
参数详解
下表详细列出了云平台 GeneActivity 分析模块的主要参数及其说明。
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头。 |
| 分组因子 | 选择要分析的细胞类型或聚类对应的标签,例如 celltype。 |
| 细胞类型 | 多选,选择要纳入分析的具体细胞类型或聚类。 |
| 分组比较 | 非必选,用于组间比较时的分组的标签,例如 Group1。 |
| 比较组 | 非必选,与分组比较参数保持一致,表示被比较的实验组。 |
| 对照组 | 非必选,与分组比较参数保持一致,表示作为对照的对照组。 |
| 拆分因子 | 多选,绘图时用于拆分不同组别的标签,如 Group 或 Sample。 |
| logFC 阈值 | 差异倍数阈值,默认 0.25。 |
| padj 阈值 | 经矫正后的显著性阈值, 默认 0.05。 |
| pval 阈值 | 显著性阈值(未进行过矫正的显著性),默认 0.05。 |
| 降维方式 | FeaturePlot 可视化 Top 差异活力的基因时用到的降维方式。例如 wnnumap。 |
重要注意事项
TIP
- 数据质量要求:计算 GeneActivity 之前需确保 fragment 文件与细胞条形码对齐,并完成基础 QC。低质量的 ATAC 数据会导致基因活力值不准确。
- 参数一致性:上游扩展距离等参数一旦确定,应在全数据分析中保持一致,以确保结果的可比性。
- 多组学数据匹配:进行跨组学比对时,确保 ATAC 和 RNA 数据来自相同的细胞或匹配的细胞群体。
操作流程
- 进入分析模块:在云平台导航至"高级分析"模块,选择"GeneActivity"。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息、上游扩展距离等参数。
- 提交任务:确认参数无误后,点击"提交"按钮,等待分析完成。
- 查看结果:分析结束后,在任务列表中查看生成的分析报告和结果文件,包括差异表格、可视化图表等。
结果解读
GeneActivity 的分析报告包含多种图表和数据文件,以下是对其核心结果的简要解读。整体结果分为两大类:基因活力差异分析,以及基因活力与基因表达的相关性分析(多组学相关性分析)。
基因活力差异分析
差异表格
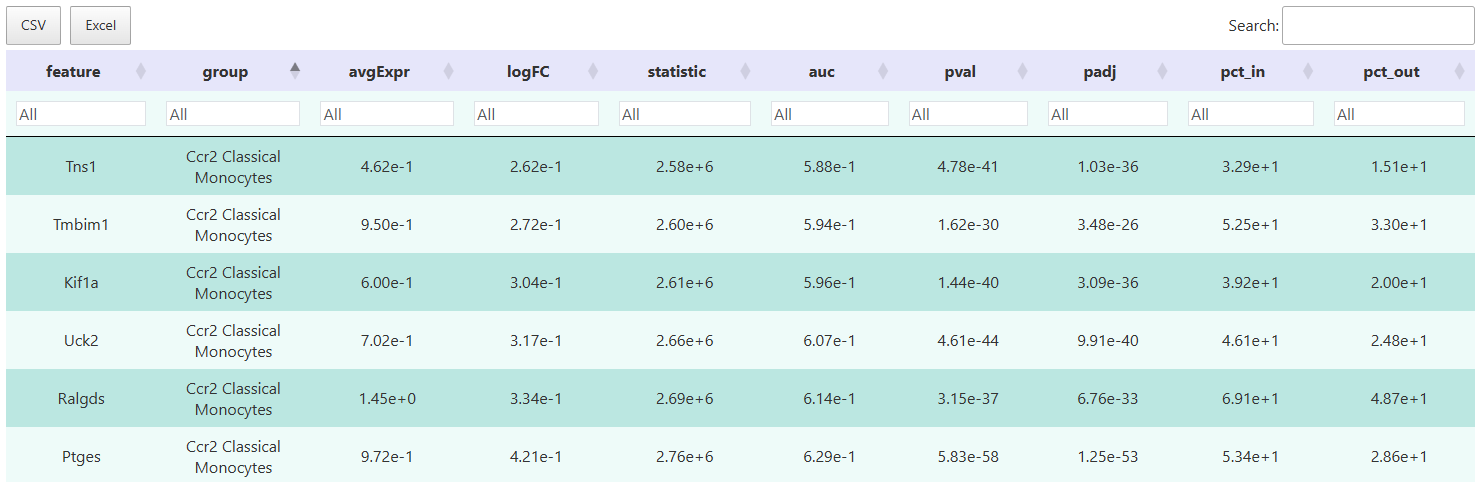
交互式表格可根据 logFC、p 值及检出率等多维指标筛选目标基因,支持导出结果,便于后续注释、功能富集分析或自定义可视化。
平台会基于 logFC Top 基因生成多种可视化结果,整合 ATAC 与 RNA 模态,辅助识别关键调控事件。
结果字段说明:
feature:基因名称。cluster/group:细胞类型或分组标签。avgExpr/logFC:平均活力和 log2FC。pct_in/pct_out:目标组和背景组的检出率。statistic、auc、p_val、p_val_adj:统计相关指标。
FeaturePlot
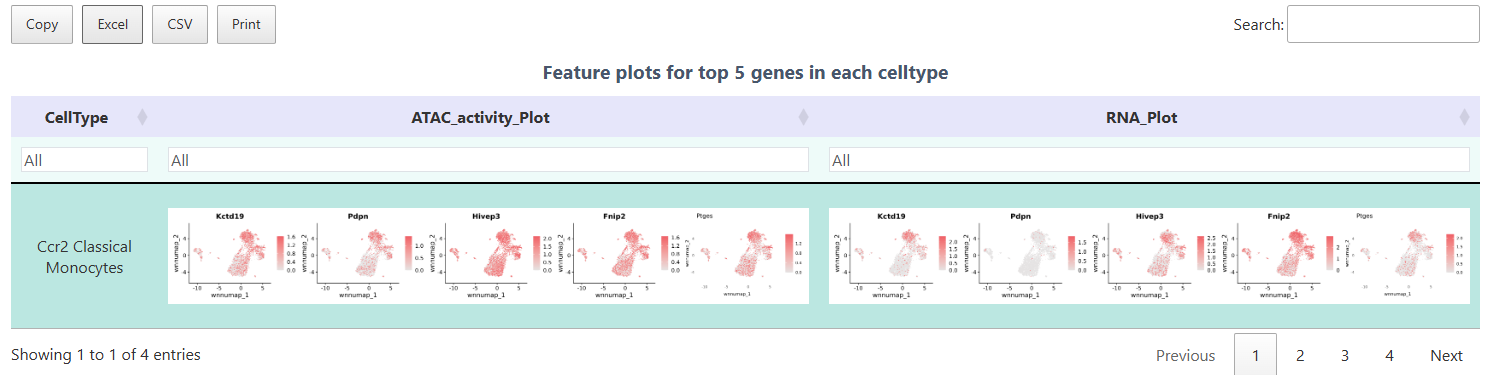
FeaturePlot 函数可同时展示 ATAC 基因活力与 RNA 表达,有助于对比染色质开放程度和转录水平。图中每个点代表一个细胞,颜色深浅分别对应目标基因在 ATAC(活力)和 RNA(表达)两层面的丰度。通过该图,可以直观验证 Top 差异基因在对应细胞群体中的表达和开放情况,从而实现多组学结果的验证。
双热图
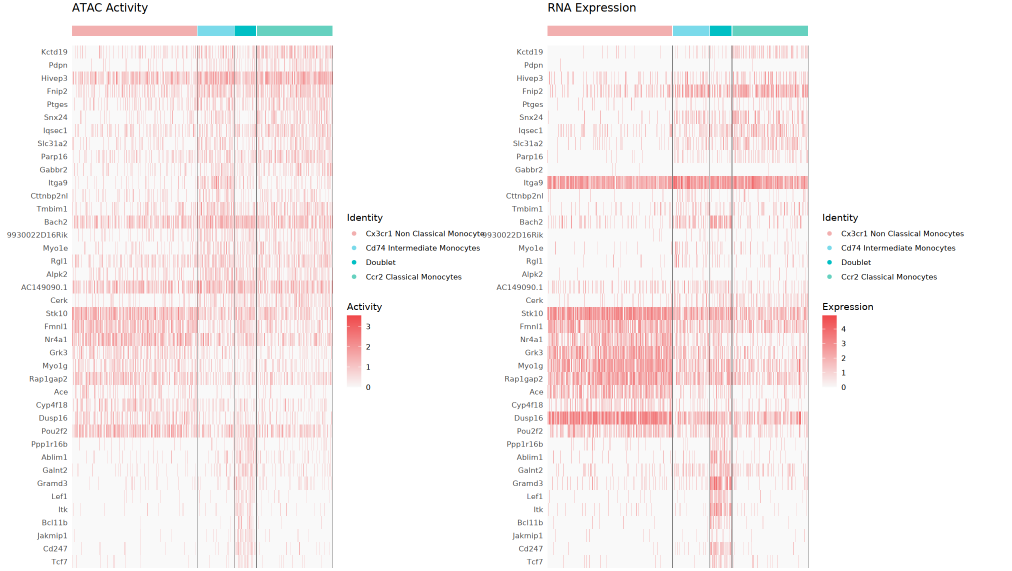
左侧为 GeneActivity(ATAC 活力)热图,右侧为 RNA 表达热图。每行对应一个 Top 差异基因,每列为单个细胞,顶端可见不同颜色标注的细胞分群。通过两者对比,可评估染色质开放和转录表达的一致性及差异,也有助于发现仅开放未表达、仅表达未开放等特殊调控现象。
小提琴图
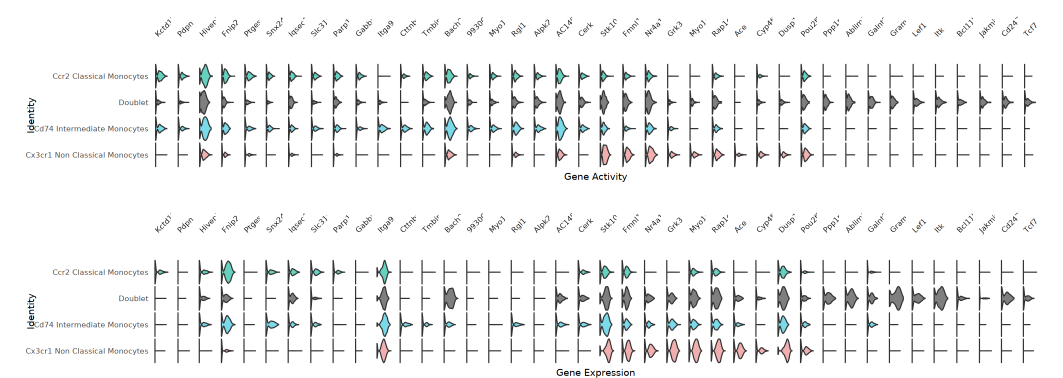
每个基因对应一组 ATAC(基因活力)和 RNA(基因表达)的小提琴图,展示不同细胞群体在这两种模态下的分布特征。该图有助于识别高活力低表达、或高表达活力不高的基因,提示潜在的调控机制。
气泡图
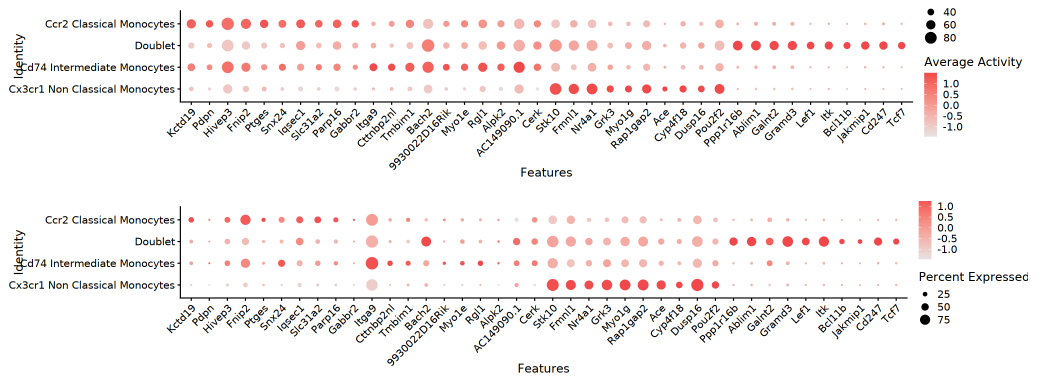
双层气泡图以气泡大小显示检出率(pct_in),颜色深浅代表平均基因活力或表达量。在细胞群体与基因的矩阵中可方便定位重要差异基因,高效显示核心调控特征。
TIP:上述可视化方法(FeaturePlot、双热图、小提琴图、气泡图)均反映"基因活力差异倍数 Top 基因"在 RNA(表达量)与 ATAC(基因活力)两个层面的分布。通过双模态对照,直观识别关键调控基因及其多组学层面的匹配关系。
多组学相关性分析
本部分的重点是比较不同细胞群(类型)在 RNA 表达与 ATAC 基因可及性两个层面的异质性和相关性。分析流程为:先分别计算每个细胞群的 RNA 平均表达量和 ATAC 基因活力均值,得到以细胞群为单位的均值矩阵;随后利用 Spearman 秩相关系数,对所有细胞群进行相关性分析,分别获得 RNA 表达和 ATAC 活力的相关性矩阵。
- RNA 相关性矩阵:用于评估细胞群间转录表达的异质性及相似性;
- ATAC 活力相关性矩阵:反映各细胞群在染色质可及性(开放状态)上的一致性或多样性。
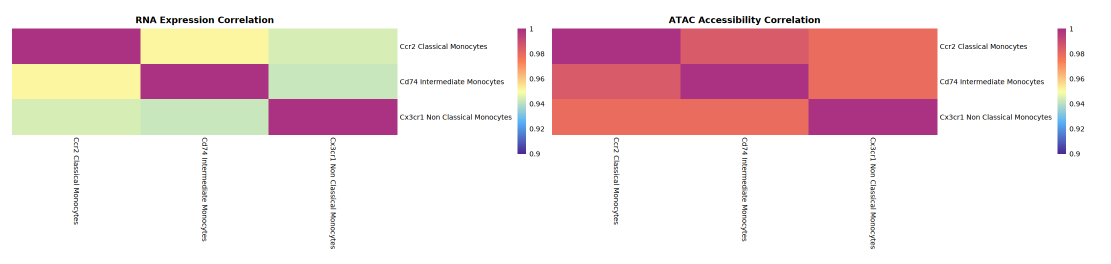
相关性热图的左侧呈现细胞群(或类型)间的 RNA 表达相关性,右侧显示 ATAC 基因活力相关性。横纵轴均为细胞群,每个方格颜色代表两者的相关系数(表达或活力相关性):颜色越红代表相关性越高,越蓝则相关性低。通过热图对比,可鉴别哪些细胞群体在 RNA 和 ATAC 层面均高度相关,哪些只在其中之一表现出相关性,从而揭示表观遗传调控与转录表达的一致或解耦关系。
解读建议:
- 利用相关性热图整体观测 ATAC 活力与 RNA 表达的相关性分布。若 ATAC 层面的相关性更高,说明表达的异质性大于基因活力异质性;反之亦然。
- 在单细胞多组学数据中,虽 ATAC peaks 开放常呈现更多异质性,但以 GeneActivity 统计的 ATAC 活力层面,其异质性通常小于 RNA 表达。具体结论需配合热图与数据进一步分析不同层面对细胞群异质性的意义。
注意事项
1. 避免过度解读:GeneActivity 值反映的是潜在的转录能力,而非实际的 mRNA 表达量。染色质开放只代表具备转录条件,仍可能受到抑制性组蛋白修饰、缺乏关键转录因子或转录后调控的影响。
2. 参数选择的重要性:上游扩展距离等参数的选择会影响基因活力的计算结果。默认的"基因体 + 上游 2 kb"范围兼顾启动子与转录延伸阶段,但可根据研究需要调整(如 1 kb / 2.5 kb),需保持全数据分析的一致性。
3. 跨组学比对需谨慎:进行 ATAC 和 RNA 数据比对时,需确保数据来自相同的细胞或匹配的细胞群体。不匹配的数据会导致错误的结论。
4. 结果不是一成不变的:GeneActivity 分析的结果会受到上游分析(如质控、标准化)以及分析参数(如上游扩展距离、差异分析阈值)的影响。如果初步结果不理想,可以尝试调整参数或重新进行上游分析。
常见问题解答 (FAQ)
Q1:GeneActivity 值为何与 RNA 表达不完全一致?
A:这是正常现象,原因包括:
- 染色质开放只代表具备转录条件,仍可能受到抑制性组蛋白修饰、缺乏关键转录因子或转录后调控的影响。
- 相反,RNA 高但 GeneActivity 低多由 ATAC 稀疏或远程增强子调控造成。
- 技术因素:ATAC 和 RNA 数据的捕获效率、测序深度等技术差异也会导致不一致。
Q2:为何默认采用"基因体 + 上游 2 kb"?可否调整?
A:
- 该范围兼顾启动子与转录延伸阶段,既具生物学意义又能缓解 ATAC 数据稀疏问题。
- 上游扩展距离参数可按需调整(如 1 kb / 2.5 kb),但需保持全数据分析的一致性。
- 不同物种或研究场景可能需要不同的扩展距离,建议参考相关文献或进行参数敏感性分析。
Q3:如何选择合适的差异分析参数?
A:
- min.pct:建议设置为 0.1-0.25,过滤掉在太少细胞中检测到的基因。
- logfc.threshold:建议设置为 0.25-0.5,根据数据质量和研究需求调整。
- p_val_adj:通常使用 0.05 作为显著性阈值,但可根据多重检验校正方法调整。
Q4:GeneActivity 分析需要哪些输入文件?
A:
- ATAC 数据:包含 fragment 信息的 Seurat 对象或 RDS 文件,已完成质控和基础分析。
- 注释信息:包含基因坐标信息的注释文件(如 GTF/GFF),用于定义基因区域。
- 细胞注释:包含细胞类型、样本分组等信息的 metadata。
Q5:如何解读双热图中的不一致现象?
A:
- 仅开放未表达:可能表示基因处于"准备转录"状态,但受到转录抑制或需要额外的激活信号。
- 仅表达未开放:可能由远程增强子调控、转录后调控或 ATAC 数据稀疏导致。
- 两者一致:表示表观遗传调控与转录表达高度一致,是典型的活跃转录基因。
Q6:GeneActivity 可以用于哪些下游分析?
A:
- 细胞类型注释:利用基因活力信息辅助细胞类型识别。
- 差异分析:识别不同细胞群体间基因活力的差异。
- 功能富集分析:对差异基因活力进行功能注释和通路富集。
- 跨组学整合:与 RNA 表达数据结合,进行多组学整合分析。
- 轨迹分析:结合拟时序分析工具,研究基因活力在细胞分化过程中的动态变化。
参考资料
[1] STUART T, SRIVASTAVA A, MADAD S, et al. Single-cell chromatin state analysis with Signac[J]. Nature methods, 2021, 18(11): 1333-1341.
[2] GRANJA J M, KLEMM S, MCGEOUGH L J, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia[J]. Nature biotechnology, 2019, 37(12): 1458-1465.
[3] BUENROSTRO J D, GIRESI P G, ZABA L C, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position[J]. Nature methods, 2013, 10(12): 1213-1218.