单细胞 scATAC-seq & scRNA-seq 双组学标准分析:Peak2Gene 分析
前言
TIP
Peak2Gene (Peak-Gene linking) 分析是一种专为单细胞多组学 (ATAC+RNA) 数据设计的方法。其核心目标是识别基因表达与其附近染色质可及性峰 (peaks) 之间的显著调控关系。
在单细胞多组学研究中,scATAC-seq 和 scRNA-seq 数据提供了不同层面的信息。Peak2Gene 分析通过统计每个细胞中基因表达量与其附近 peaks 的 ATAC 信号强度间的相关性,并利用广义线性模型校正 GC 含量、peak 长度、距离等技术偏置,从而推断哪个 peak 可能参与调控哪些基因。Peak2Gene 分析不仅帮助揭示顺式调控网络(如增强子-基因连接),还能与 motif 富集/活性结果结合,系统性挖掘关键转录因子的直接靶基因和调控作用,是多组学调控机制研究的基础工具之一。
Peak2Gene 的核心功能
peak-gene 关联识别 通过计算基因表达量与其邻近 peaks 的可及性信号之间的相关性,识别可能调控该基因的 peak 集合,克服仅靠线性距离注释的局限。
技术偏置校正 使用广义线性模型校正 GC 含量、整体可及性、peak 长度等多种技术偏置,获得更为可靠的 peak-gene 关联。
顺式调控网络构建 帮助揭示顺式调控网络(如增强子-基因连接),为多组学调控机制研究提供基础。
与 Motif 分析整合 结合 motif 富集/活性结果,系统性挖掘关键转录因子的直接靶基因和调控作用,构建完整的 TF → peak → gene 调控轴。
本篇文档旨在为单细胞多组学研究者提供一份详尽的 Peak2Gene 技术指南,内容涵盖其基本原理、在 SeekSoul Online 云平台上的操作方法、结果解读及常见问题,帮助您快速掌握并应用该工具。
Peak2Gene 理论基础
核心原理
Peak2Gene 分析的核心思想是:通过统计每个细胞中基因表达量与其附近 peaks 的 ATAC 信号强度间的相关性,并利用广义线性模型校正多种技术偏置,从而推断哪个 peak 可能参与调控哪些基因。
基因调控网络简介
基因调控网络 (Gene Regulatory Network, GRN) 由转录因子 (TF)、调控元件(如启动子、增强子、peaks)以及靶基因构成。TF 通过识别特定 DNA motif 结合到开放染色质区域,进而影响下游基因的转录。单细胞多组学手段的优势在于,可以在每个细胞分辨率下同时观测 TF 和靶基因的表达 (scRNA-seq)、以及 TF 结合位点的染色质可及性 (scATAC-seq,并结合 motif 识别进行推断)。
通过联合单细胞 ATAC 与 RNA 数据,可实现:
- TF 表达谱的确定
- TF 结合位点 (peak) 的开放性评估
- motif 的富集情况及功能活性推断
- 靶基因表达的动态变化监测
这种 "TF 表达 × motif-peak 结合 × 靶基因表达" 三维整合,有助于重建单细胞尺度的调控因果链,精准捕捉关键调控及作用靶点,并为 GRN 建图和机制深入提供着力点。
Peak2Gene 分析如何助力解析 GRN 基因调控网络
Motif 分析:通过与已知转录因子识别序列比对,从开放染色质 peak 中识别可能被 TF 结合的区域,是精准描述 TF → peak 调控关系的重要基础。这些 motif 富集的 peaks 被视为 TF 调控潜在靶点,是调控网络的“上游”基础。
Peak2Gene 分析:通过在单细胞水平考察 peak 可及信号与附近基因表达的相关性,精确捕捉每个 peak 与其潜在靶基因之间的调控配对,克服仅靠线性距离注释的局限。这一策略有力刻画了开放 peaks 实际参与基因调控的真实证据,阐释调控网络“下游”环节。
整合分析:结合 motif 分析 (TF → peak) 与 Peak2Gene 分析 (peak → gene),能够系统性描绘 TF、调控元件 peak 及其作用靶基因三者间的直接调控轴,为多组学调控机制研究提供多重证据支撑。
TIP
标准分析流程
- 差异 peak 筛选:定位具有生物学意义的开放区域
- Peak2Gene 注释:整合表达及距离,建立 peak–gene 关联
- Motif 分析:鉴定及评估 motif 富集和活性,预测关键 TF
- 多组学联合验证:分析 motif 活性、TF/靶基因表达和 peak 可及性之间的调控一致性
关键算法与流程
对于每个目标基因,Peak2Gene 分析包括以下关键步骤:
- 筛选邻近 peaks:筛选距离该基因(如 ±500 kb)范围内的 peaks
- 计算相关性:计算这些 peaks 可及性与基因表达的相关性
- 偏置校正:使用广义线性模型校正 GC%、peak size、整体可及性等偏置,获得更为可靠的 peak-gene 关联
这种方式可以精确定量每个 peak 对靶基因的调控支持证据,是重构多组学调控网络的关键环节。实际项目落地时,推荐对感兴趣的基因集先行评估,也可扩展至全基因组自动注释。
云平台操作指南
在云平台上,Peak2Gene 分析流程被设计得直观易用。您无需编写代码,只需通过参数配置界面即可完成分析。
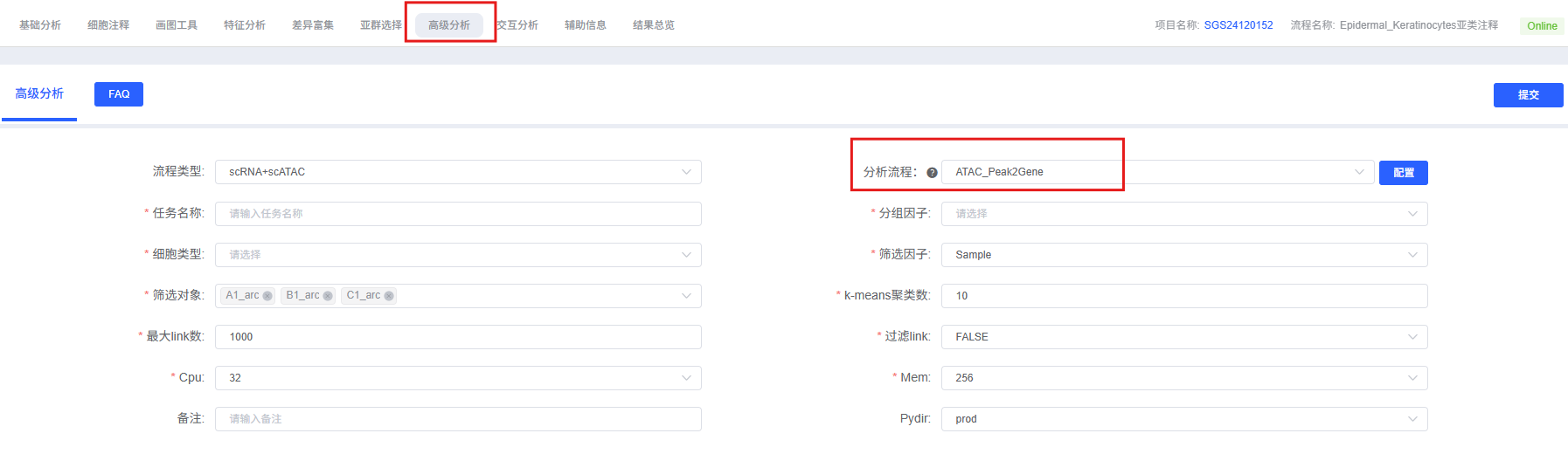
分析前的准备
TIP
Peak2Gene 分析的成功与否,很大程度上取决于输入数据的质量和多组学数据的匹配度。在开始分析前,请务必确认:
- 数据已完成预处理:您的单细胞 ATAC 和 RNA 数据已经过标准的质控、降维、聚类和细胞类型注释。
- 数据匹配性:确保 ATAC 和 RNA 数据来自相同的细胞或匹配的细胞群体,以便进行相关性分析。
- 注释信息完整:确保数据中包含必要的细胞类型、样本分组等注释信息,以便进行结果展示和解读。
- RegionStats 步骤:确保已执行 RegionStats 步骤,注释每个 peak 区域的 GC 含量等特征,用于后续偏置校正。
参数详解
下表详细列出了云平台 Peak2Gene 分析模块的主要参数及其说明。
| 参数名称 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头,可包含英文字母、数字、下划线和中文。 |
| 分组因子 | 细胞分群列名。 |
| 细胞类型 | 要分析的细胞类型。 |
| 筛选因子 | 要分析的样本名称或分组名。 |
| 筛选对象 | 要分析的样本名称或分组名。 |
| k-means 聚类数 | K-means 聚类的簇数,用于 link 分组,默认为 10。 |
| 最大 link 数 | 绘制的最大 link 数量,默认为 5000。 |
| 过滤 link | 选项 TRUE or FALSE,若为 TRUE 则显示以下四个参数。 |
| 相关性阈值 | 相关性阈值,用于过滤 link,默认为 0.1。 |
| p 值阈值 | P 值阈值,用于统计显著性过滤,默认为 0.01。 |
| ATAC 方差阈值 | ATAC 数据的方差阈值,用于过滤低变异性 peak,默认为 0.25。 |
| RNA 方差阈值 | RNA 数据的方差阈值,用于过滤低变异性 gene,默认为 0.25。 |
重要注意事项
TIP
- 数据质量要求:确保 ATAC 和 RNA 数据的质量满足要求,低质量数据会导致 peak-gene 关联不准确。
- 数据匹配性:进行相关性分析时,需确保 ATAC 和 RNA 数据来自相同的细胞或匹配的细胞群体。
- 距离阈值选择:默认 500 kb 的距离阈值适用于大多数情况,可根据研究需要调整。
- 显著性阈值:建议使用默认的 p 值阈值 (0.05),过严格的阈值可能会过滤掉较多关联。
- 与 Motif 分析整合:Peak2Gene 分析结果可以与 Motif 分析结果整合,构建完整的 TF → peak → gene 调控轴。
操作流程
- 进入分析模块:在云平台导航至“高级分析”模块,选择 "Peak2Gene"。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息、目标基因等参数。
- 提交任务:确认参数无误后,点击“提交”按钮,等待分析完成。
- 查看结果:分析结束后,在任务列表中查看生成的分析报告和结果文件,包括 peak-gene 关联热图、相关性分析结果等。
结果解读
Peak2Gene 的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
Peak-Gene 相关性热图
热图展示了染色质开放区域 (peaks) 与基因表达之间的相关性模式:
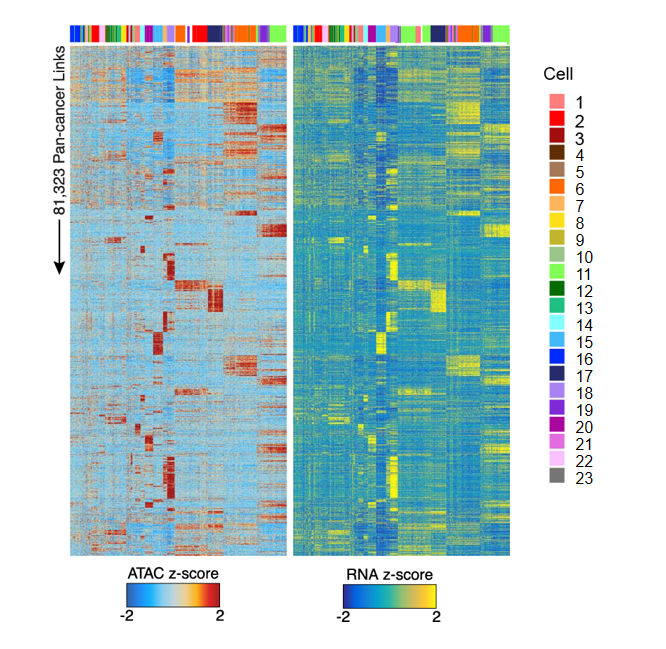
图表解读
热图分为两部分:
- ATAC 热图(左):显示 ATAC-seq 信号强度(染色质可及性),蓝色表示低可及性,红色表示高可及性。
- RNA 热图(右):显示相应基因的表达水平,蓝色表示低表达,黄色表示高表达。
- 每一行代表一个 peak-gene 对,每一列代表一个单细胞。
- 顶部颜色条标识不同细胞类型或群体,便于观察特定细胞群体中的 peak-gene 关联模式。
分析要点
- 关联模式识别:通过观察热图中 ATAC 和 RNA 信号的一致性,可以识别 peak-gene 关联的模式。
- 细胞类型特异性:不同细胞类型可能表现出不同的 peak-gene 关联模式,反映了细胞类型特异性的调控机制。
- 调控方向判断:通过比较 peak 可及性和基因表达的相关性,可以判断调控的方向和强度。
关联 peak 数分布直方图
该直方图展示了每个基因关联到 1~25 个 peak 的数量分布:
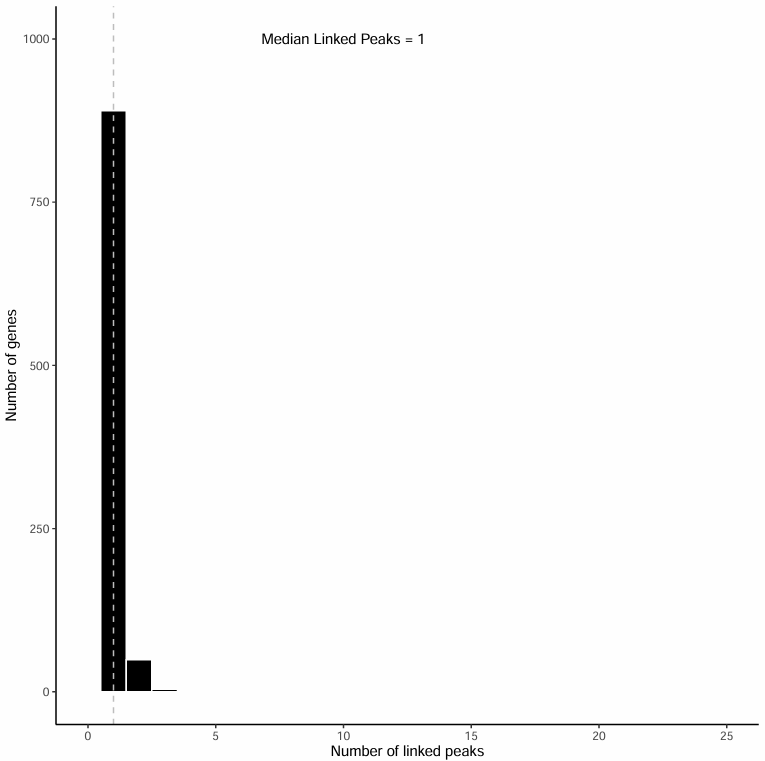
图表解读
- 横轴:基因所关联的 peak 数量 (1~25)
- 纵轴:具有相应关联 peak 数的基因数量
- 虚线表示中位数位置,并以文本注明具体中位数
分析要点
- 关联数量分布:通过观察直方图,可以了解大多数基因关联的 peak 数量分布情况。
- 中位数评估:中位数反映了基因关联 peak 数量的典型水平,有助于评估分析的质量。
- 异常值识别:关联 peak 数量异常多或异常少的基因值得进一步关注。
Top 显著 peak-gene 关联火山图
火山图聚焦于 Top 10 最显著的 peak-gene 关联:
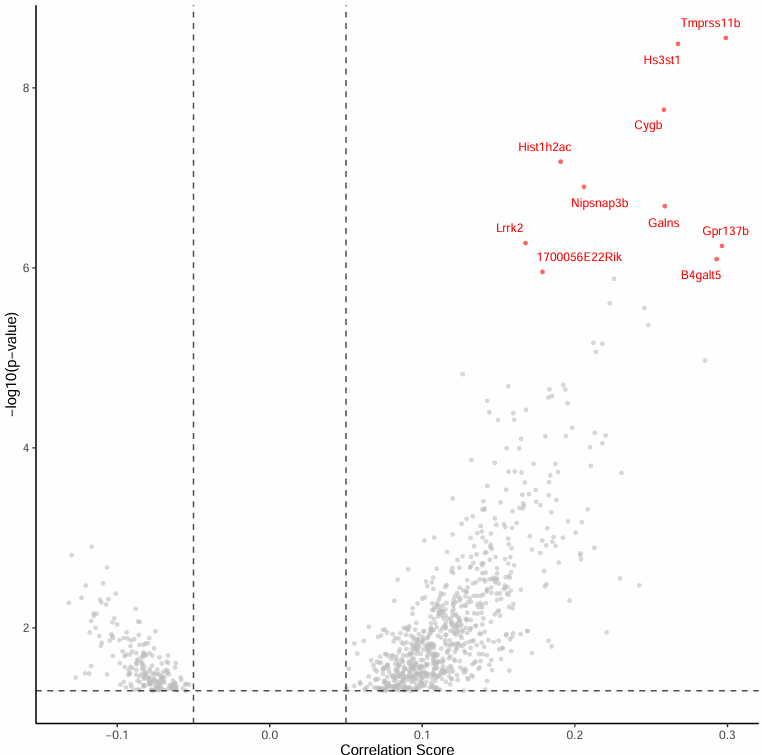
图表解读
- 横轴:相关性 score
- 纵轴:-log10(p) 值
- 标注 Top 10 显著基因便于直观呈现
分析要点
- 显著关联识别:Top 10 最显著的 peak-gene 关联是研究的重点,值得进一步分析。
- 相关性强度:通过观察相关性 score,可以评估 peak-gene 关联的强度。
- 统计显著性:通过观察 -log10(p) 值,可以评估关联的统计显著性。
全部 peak-gene 对相关性分布曲线
曲线图整体展示了全部 peak-gene 对的相关系数分布:
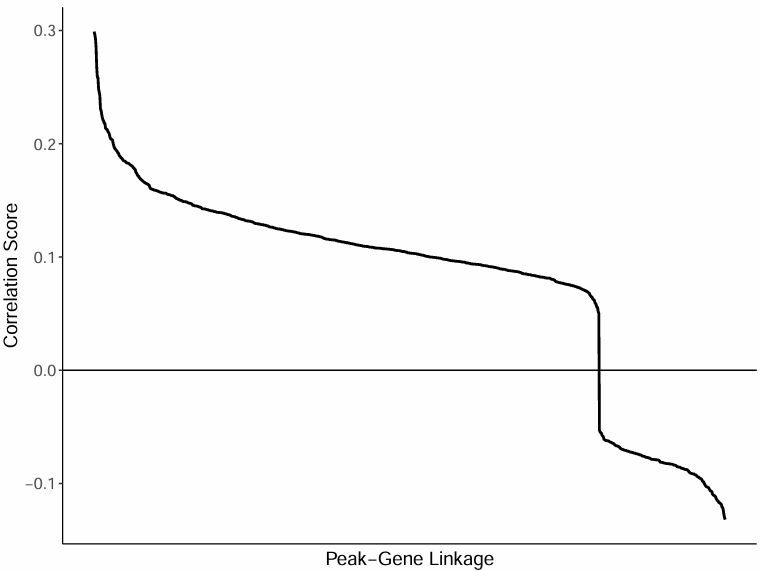
图表解读
- 横轴:peak-gene 对(按相关性高到低排序)
- 纵轴:对应相关系数
分析要点
- 整体分布模式:通过观察曲线图,可以了解全部 peak-gene 对的相关性分布模式。
- 强关联识别:相关性强的 peak-gene 对位于曲线的左侧,是研究的重点。
- 阈值选择:可以根据曲线图选择合适的相关性阈值,用于筛选显著的关联。
结果文件列表
| 文件名 | 内容说明 |
|---|---|
peak_gene_links.csv | 核心数据:包含所有显著的 Peak-to-Gene 关联及其相关性评分和 p 值。 |
Peak2GeneHeatmap.pdf/png | Peak-to-Gene 关联热图。 |
Peak2GeneHistogram.pdf/png | Peak-to-Gene 直方图。 |
Peak2GeneVolcano.pdf/png | Peak-to-Gene 小提琴图。 |
Peak2GeneRank.pdf/png | Peak-to-Gene 排序曲线图。 |
注意事项
1. 数据质量的重要性:Peak2Gene 分析的结果质量很大程度上取决于输入数据的质量。确保 ATAC 和 RNA 数据都经过充分的质控和标准化处理。
2. 数据匹配性:进行相关性分析时,需确保 ATAC 和 RNA 数据来自相同的细胞或匹配的细胞群体。不匹配的数据会导致错误的关联。
3. 距离阈值的选择:默认 500 kb 的距离阈值适用于大多数情况,但可根据研究需要调整。较小的阈值可能遗漏远程调控元件,较大的阈值可能引入噪音。
4. 显著性阈值:建议使用默认的 p 值阈值 (0.05),过严格的阈值可能会过滤掉较多关联,过宽松的阈值可能引入假阳性。
5. 与 Motif 分析整合:Peak2Gene 分析结果可以与 Motif 分析结果整合,构建完整的 TF → peak → gene 调控轴。建议在分析流程中同时进行这两种分析。
6. 结果解读需谨慎:Peak2Gene 分析识别的是相关性,而非因果关系。结果需要结合生物学知识和其他证据进行解读和验证。
常见问题解答 (FAQ)
Q1:Peak2Gene 分析与 Motif 富集有什么联系?
A:两者关注角度不同,但结果可互补验证:
- Peak2Gene:主要回答“哪些 peak 实际调控了哪些基因”,通过相关性判定 peak-gene 调控关系,输出为 peak-gene 配对及相关性评分。
- Motif 富集:聚焦于“某一组 peak 中富集了哪些转录因子结合位点 (motif)”,反映哪些 TF 可能调控这些 peak,输出为 motif 丰度及统计显著性。
- 整合分析:结合 motif 分析 (TF → peak) 与 Peak2Gene 分析 (peak → gene),能够系统性描绘 TF、调控元件 peak 及其作用靶基因三者间的直接调控轴。
Q2:为什么 Peak2Gene 得到的关联数较少?
A:可从以下方面排查原因:
- 样本或细胞数量较少:导致统计功效不足,建议增加样本或细胞数量
- 选取的基因集过小:或目标区域受限,建议扩大分析范围或进行全基因组分析
- 设定的显著性阈值过高:过滤掉了较多关联,建议适当放宽阈值
- RegionStats 步骤未正确执行:缺少偏置校正信息,建议确保已正确执行 RegionStats 步骤
- 数据匹配性问题:Motif 富集与 Peak2Gene 分析使用了不同细胞子集或过滤条件,建议使用相同的细胞子集
Q3:如何选择合适的距离阈值?
A:距离阈值的选择需要平衡灵敏度和特异性:
- 默认 500 kb:适用于大多数情况,能够捕获大多数顺式调控元件
- 较小阈值(如 100 kb):更严格,可能遗漏远程调控元件,但能减少噪音
- 较大阈值(如 1 Mb):更宽松,可能捕获更多远程调控元件,但可能引入噪音
- 建议:可以先使用默认阈值,然后根据结果质量进行调整
Q4:Peak2Gene 分析可以用于哪些下游分析?
A:Peak2Gene 分析结果可以用于多种下游分析:
- 差异 peak 分析:结合差异 peak 分析结果,识别具有生物学意义的 peak-gene 关联
- Motif 分析整合:与 Motif 分析结果整合,构建完整的 TF → peak → gene 调控轴
- 功能富集分析:对关联的基因进行功能富集分析,揭示调控的生物学功能
- 多组学整合:与 scRNA-seq 数据整合,揭示染色质可及性与基因表达的时序关联
- 调控网络构建:基于 peak-gene 关联构建基因调控网络
Q5:如何判断 Peak2Gene 分析的质量?
A:判断 Peak2Gene 分析质量的几个关键指标:
- 关联数量:合理的关联数量反映了分析的统计功效
- 相关性分布:相关性分布应该呈现合理的模式,强关联应该占一定比例
- 生物学合理性:关联的 peak-gene 对应该符合已知的生物学知识
- 细胞类型特异性:不同细胞类型应该表现出不同的 peak-gene 关联模式
Q6:Peak2Gene 分析与传统的距离注释有什么区别?
A:Peak2Gene 分析相比传统的距离注释有以下优势:
- 相关性证据:基于单细胞水平的相关性分析,提供更可靠的调控证据
- 技术偏置校正:使用广义线性模型校正多种技术偏置,提高准确性
- 细胞类型特异性:能够识别细胞类型特异性的 peak-gene 关联
- 动态变化:能够揭示 peak-gene 关联在不同细胞状态下的动态变化
参考资料
[1] STUART T, SRIVASTAVA A, MADAD S, et al. Single-cell chromatin state analysis with Signac[J]. Nature methods, 2021, 18(11): 1333-1341.
[2] GRANJA J M, KLEMM S, MCGEOUGH L J, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia[J]. Nature biotechnology, 2019, 37(12): 1458-1465.
[3] PLINER H A, PACKER J S, MCFALINE-FIGUEROA J L, et al. Cicero predicts cis-regulatory DNA interactions from single-cell chromatin accessibility data[J]. Molecular cell, 2018, 71(5): 858-871.