LncCoExpression 分析
前言
TIP
LncCoExpression 模块面向单细胞 RNA 测序(scRNA-seq)数据,旨在刻画 lncRNA 与蛋白编码基因(protein-coding RNA)的共表达关系,构建细胞群特异的共表达网络与功能模块,并对模块进行功能富集解读,用于挖掘关键 lncRNA 及其潜在作用通路。
长链非编码 RNA(lncRNA)是长度超过 200 nt 的非编码转录本,在染色质状态维持、转录调控、细胞分化与疾病进展中发挥广泛作用。基于单细胞分辨率的 lncRNA 表达谱,我们可与蛋白编码基因一起构建共表达网络;在网络中再通过图社区划分提取功能模块,并结合 GO/KEGG 等通路进行生物学解释。
LncCoExpression理论基础
核心原理
- 差异基因获取:以细胞簇(cluster)或分组比较为粒度,使用 Seurat 的 FindAllMarkers/FindMarkers 获取差异表达的 lncRNA 与蛋白编码基因(默认阈值:|avg_log2FC| ≥ 0.25,p_val_adj < 0.05)。
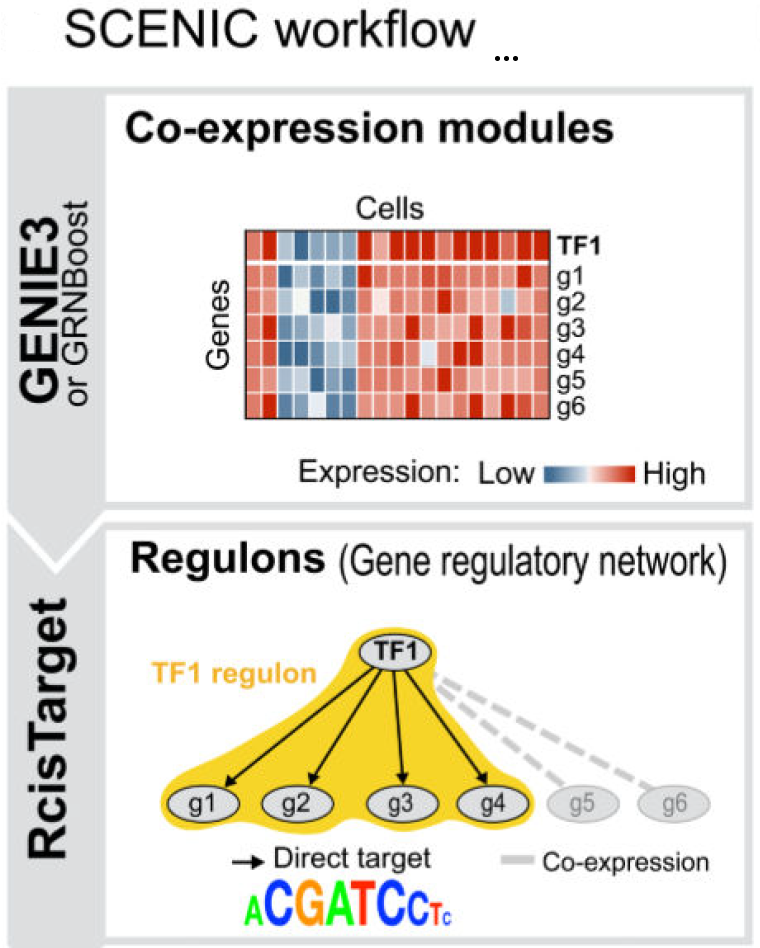
- 共表达网络推断:对保留下来的差异基因集合,采用 pySCENIC 工作流中的 Grnboost2 进行基因间共表达关系学习,得到每条边的 importance(权重)。
- 网络过滤与建图:按 importance 从高到低保留 Top 0.5% 的边,使用 networkx 构图,并要求每个 protein-coding 基因至少与一个 lncRNA 相连,以确保网络围绕 lncRNA 展开。
- 模块划分与注释:利用 Louvain 算法对网络做社区检测,过滤掉 degree < 7 的小模块,剩余模块进入功能富集分析(clusterProfiler,GO/KEGG)。
TIP
以上流程会分别在「每个细胞簇」与「每个分组比较」中独立执行,从而得到 cluster 特异与条件对比特异的 lncRNA 共表达网络与功能模块。
原理示意
云平台操作指南
在云平台上,LncCoExpression 分析被设计为可视化参数配置与一键计算。
分析前的准备
TIP
- 数据预处理就绪:建议上游已完成标准的质控、降维、聚类与细胞类型注释(如
CellAnnotation)。 - 注释列名规范:元数据(metadata)列名与内容建议使用英文/数字/下划线,避免中文与特殊字符。
- novel-lnc 检测:如需从比对结果识别 novel-lncRNA,请在平台参数中开启“识别 novel-lnc”,并在样本信息表提供
bam_path。该流程对 BAM 质量、参考注释完备度敏感,计算耗时较长;建议在明确研究需求时再开启。
参数详解
下表列出云平台 LncCoExpression 模块的主要参数与说明(与历史文档风格一致)。
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头,可包含英文字母、数字、下划线和中文。 |
| 物种 | 选择您的数据对应的物种:human/mouse。 |
| 识别 novel-lnc | 是否需要从比对结果中检测 novel-lncRNA。默认 FALSE。选择 TRUE 时需提供 bam_path(见“样本信息”),且该步骤耗时较久。 |
| 样本信息 | 配置参与分析的样本及其 bam 文件路径。 |
a. Sample | 选择需要分析的样本,对应 metadata 中的 Sample 或 raw_Sample 列。 |
b. bam_path | 样本对应的 BAM 数据路径,开启“识别 novel-lnc”时必填。 |
c. 操作 | 点击“删除”删除该行,点击左上角“添加”新增一行。 |
| 分组因子 | 选择要分析的细胞类型或聚类标签(如 CellAnnotation),与“细胞类型”配合使用。 |
| 细胞类型 | 多选,选择要纳入分析的具体细胞类型/聚类(如 T cell、NK cell、Monocyte 等)。 |
| 分组比较 | 定义需要进行比较分析的组合信息。 |
a. 拆分因子 | 用于定义比较组合的标签(如 Group、Sample 等)。 |
b. 处理组 | 处理组名。 |
c. 对照组 | 对照组名。 |
d. 操作 | 点击“删除”删除该行,点击左上角“添加”新增一行。 |
| 备注 | 自定义备注信息。 |
操作流程
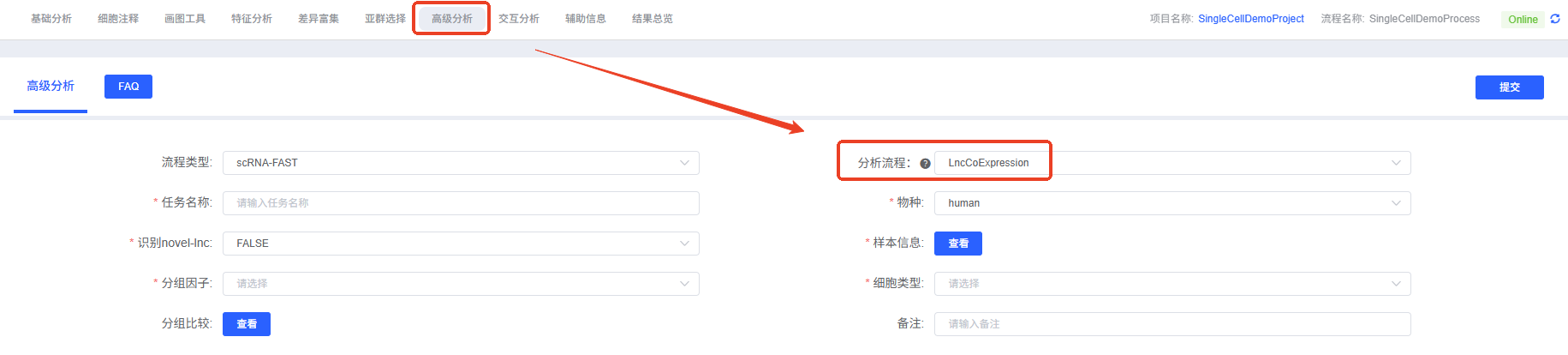
- 进入分析模块:在云平台“高级分析”中选择“LncCoExpression”。
- 创建任务:填写“任务名称”,选择“物种”。
- 配置输入:上传/选择“样本信息”,如需检测 novel-lnc,则提供
bam_path并开启对应参数。 - 定义对象:设置“分组因子”和“细胞类型”;如需比较,配置“分组比较”。
- 提交与等待:确认参数后提交任务,等待计算完成。
- 下载与查看:在任务列表下载报告与结果文件,或在页面直接预览图表。
结果解读
LncCoExpression的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
5.1 lncRNA 表达概览
5.1.1 细胞簇/样本分布 (UMAP)
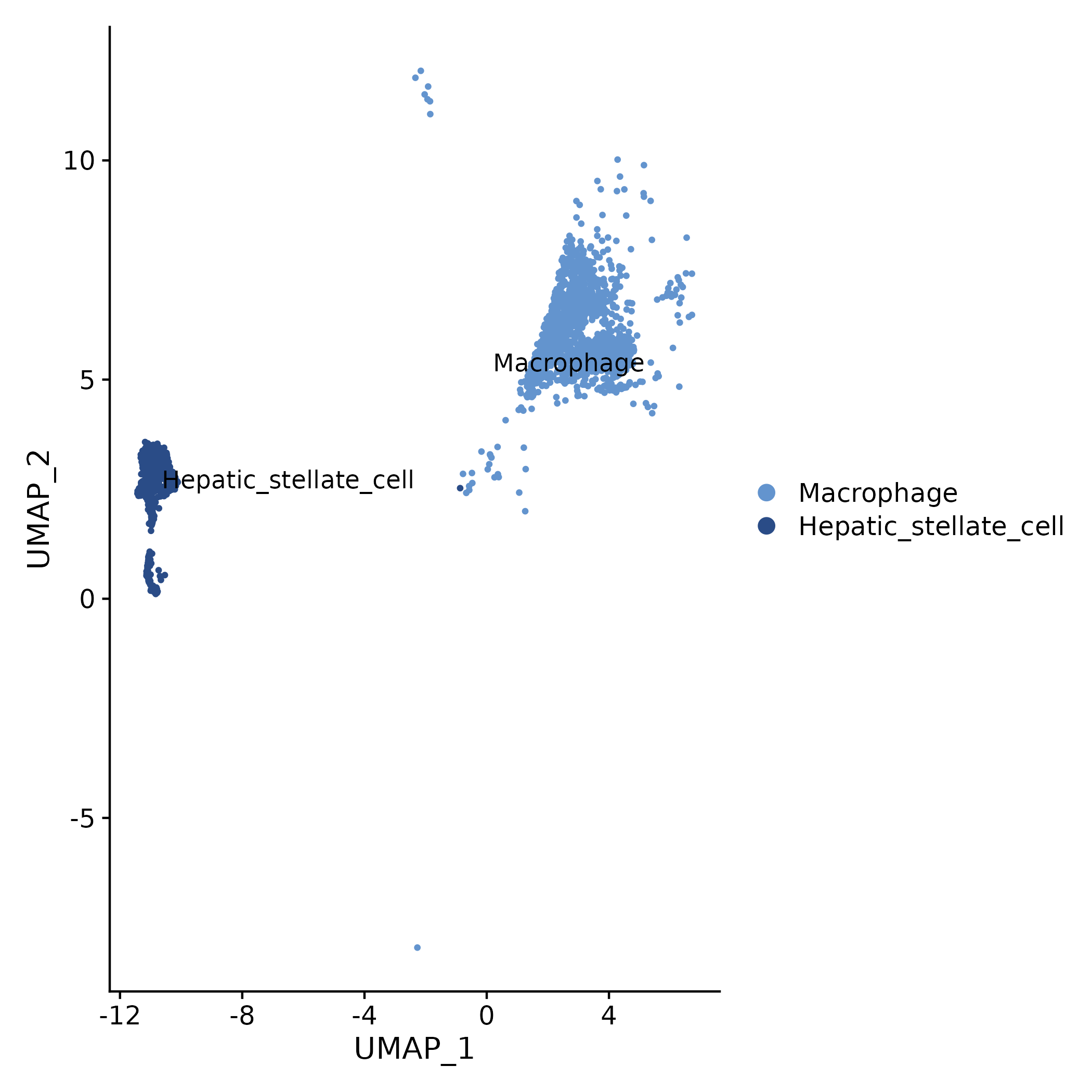
- 图表解读:每个点为细胞;不同颜色代表不同 cluster 或 sample 分组。
- 分析要点:可初步查看不同细胞群(或样本)中 lncRNA 分析的背景构成与分布差异。
5.1.2 各 cluster 的 lncRNA 表达占比
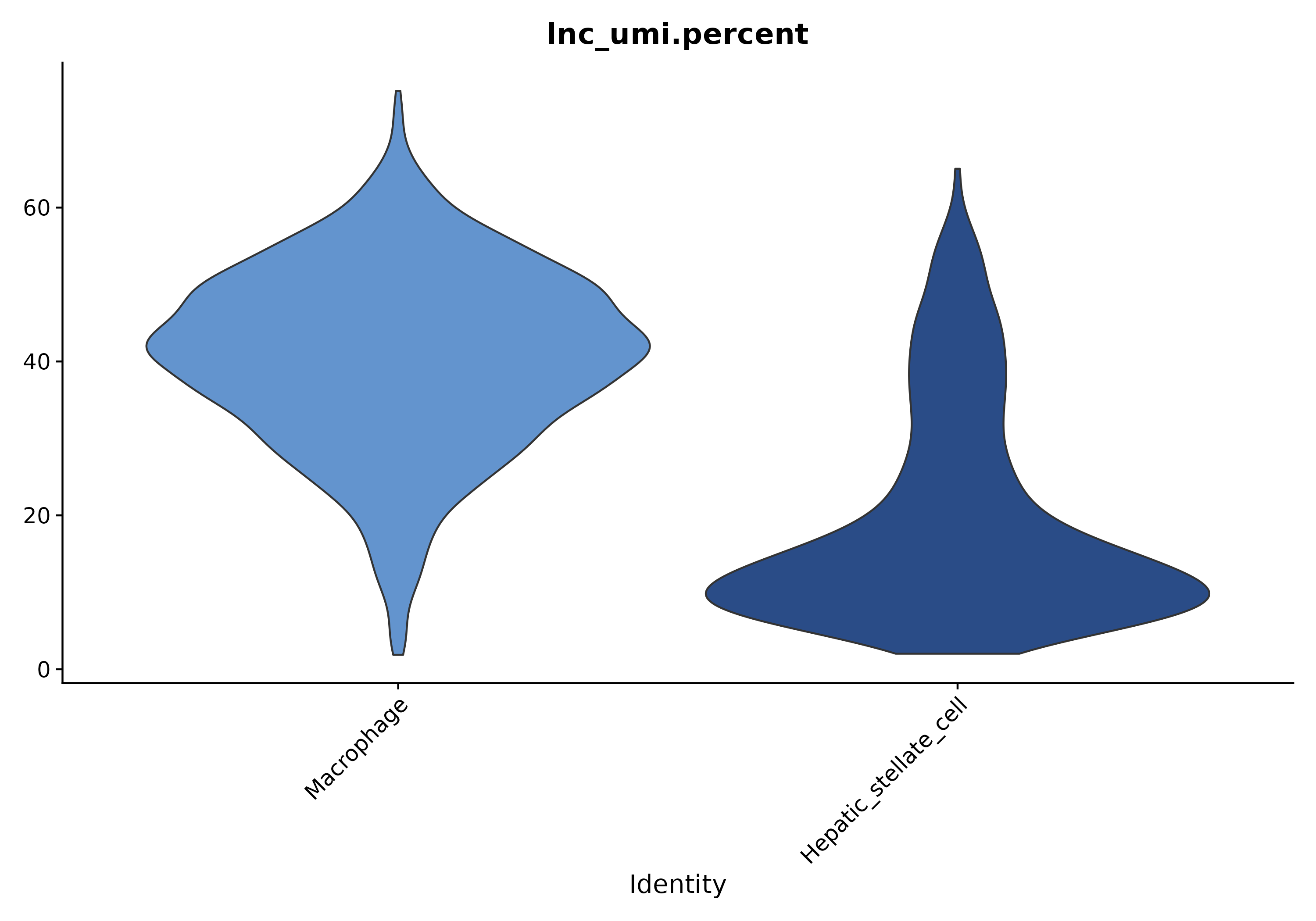
- 图表解读:横轴为细胞簇,纵轴为该簇中 lncRNA UMI 总和占全部 UMI 的比例。
- 分析要点:用于评估不同细胞簇中 lncRNA 表达总体水平差异。
5.1.3 lncRNA / protein-coding 的 nCount/nFeature
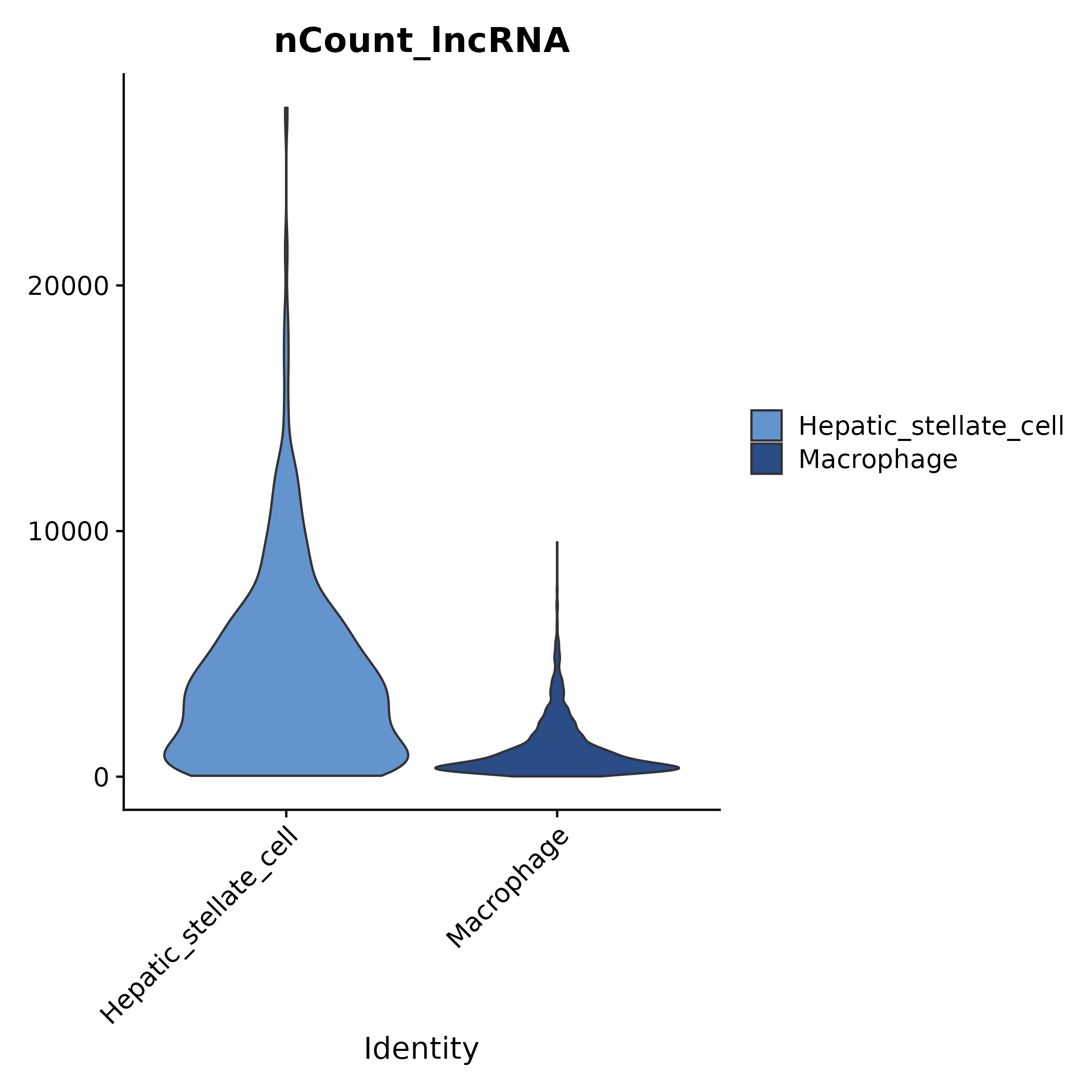
- 图表解读:展示各细胞簇在 lncRNA 与 protein-coding RNA 两类上 UMI(nCount)与基因数(nFeature)的分布。
- 分析要点:辅助判断不同簇的测序深度、检测到的基因数等对后续差异与共表达分析的潜在影响。
5.2 差异表达基因 (DEG)
- 方法说明:基于 Seurat,默认阈值为
avg_log2FC ≥ 0.25且p_val_adj < 0.05。差异集合包含 lncRNA 和 protein-coding 基因。
5.2.1 差异基因热图
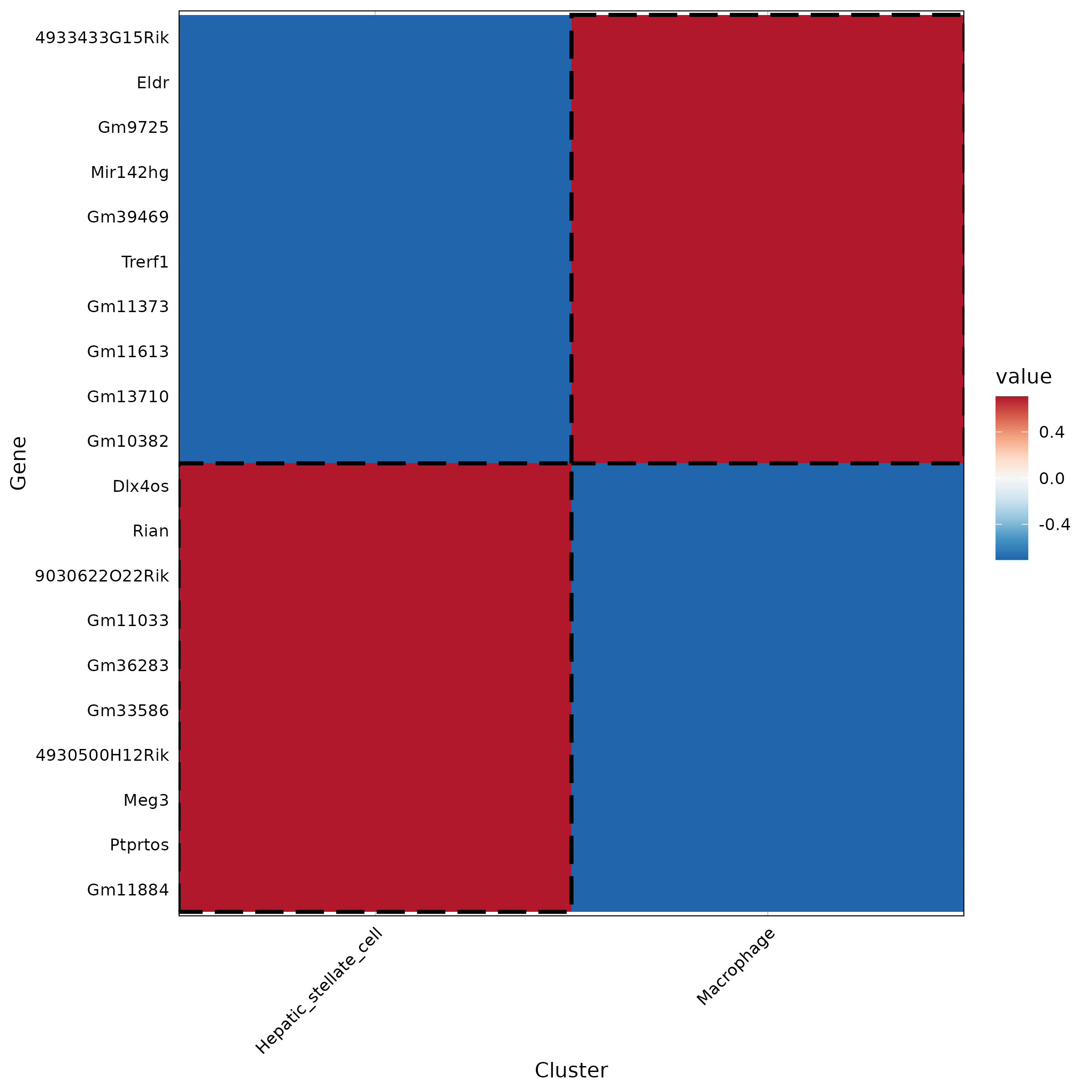
- 图表解读:每列代表一个细胞簇(或分组),展示该簇的 Top10 差异基因(lncRNA 或蛋白编码基因)在各簇的平均表达水平,颜色通常为标准化表达(如 Z-score),由蓝到红表示由低到高。
- 分析要点:定位每个簇的标志性 lncRNA 及其共表达的蛋白编码基因,关注在目标簇内高表达而在其他簇低表达的基因;这些候选集合将直接影响后续网络与功能模块的构建与解读。
5.2.2 特异性 (Tau) 评分
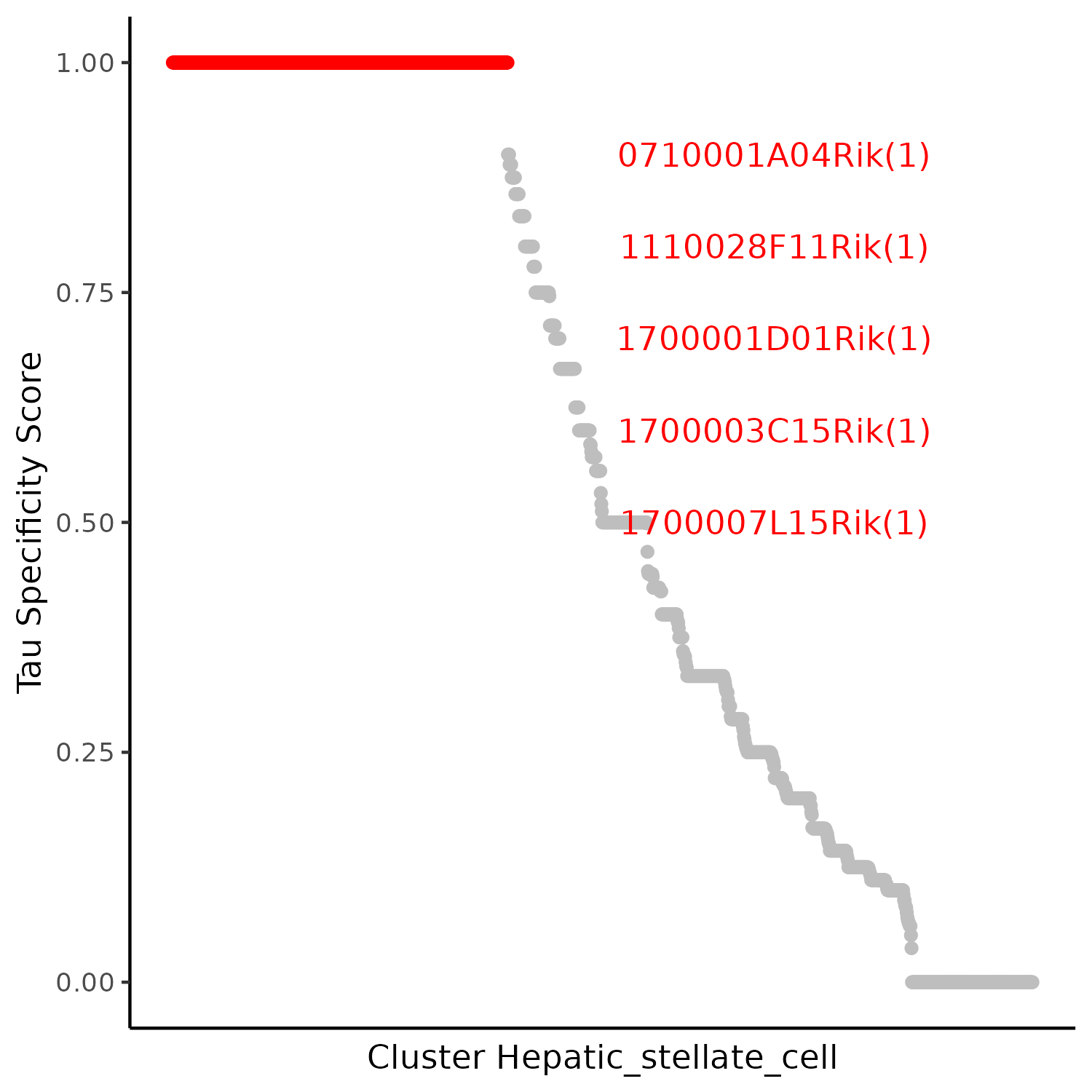
- 图表解读:每个点代表一个 lncRNA ,Tau 取值范围为 [0, 1],数值越接近 1 表明该 lncRNA 在更少的簇中高度特异表达,越接近 0 表明其在多簇中广泛表达。
- 分析要点:优先关注 Tau 高且统计显著(或具有较高表达量/效应量)的 lncRNA 作为候选调控因子;结合差异分析与网络中心性指标综合筛选更稳健。
5.2.3 单基因可视化 (示例簇 Top4 lncRNA)
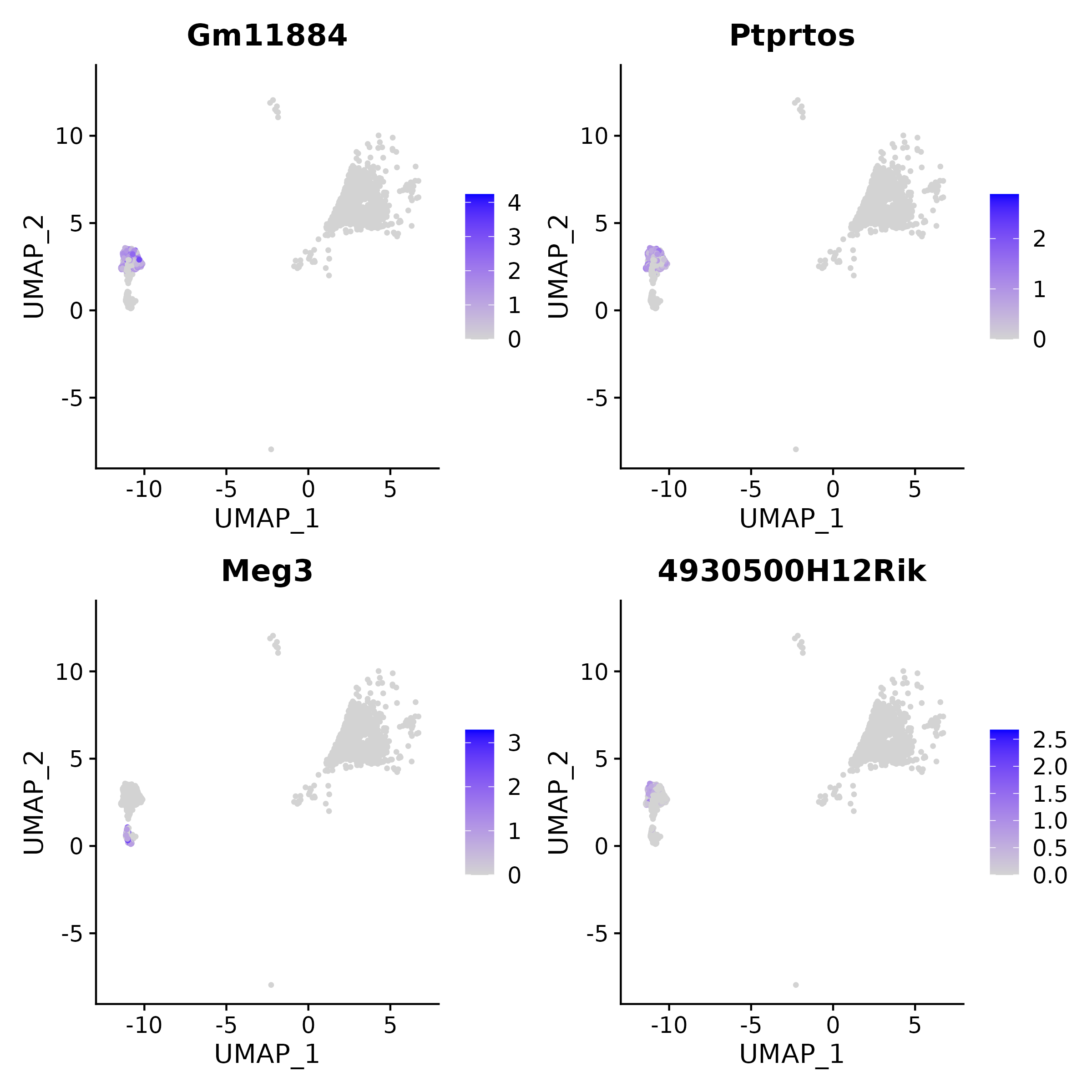
- 图表解读:在 UMAP 空间中展示单个 lncRNA 的表达分布,颜色深浅代表表达强弱,蓝色越深表示该差异 lncRNA 在细胞簇中表达更高,能直观显示其在不同细胞簇/空间位置的富集情况。
- 分析要点:用于验证候选 lncRNA 的空间/簇特异性,与细胞类型注释进行交叉验证;如出现局灶性高表达且与功能相关簇一致,更支持其为关键分子。
TIP
通过差异与特异性结合,可快速定位高特异表达的候选 lncRNA,为下游网络与机制研究提供靶点集合。
5.3 lncRNA 共表达网络 (按簇)
5.3.1 网络图
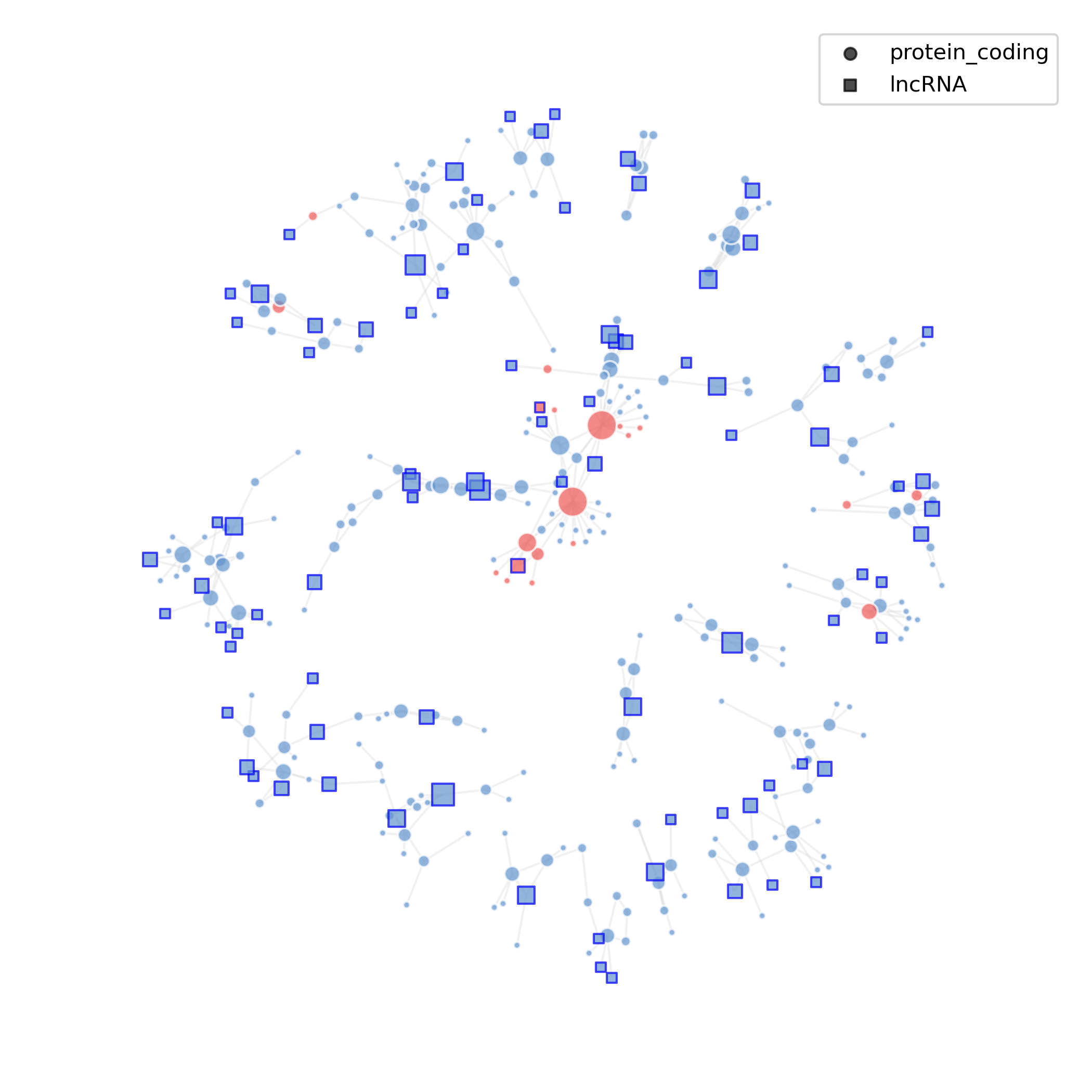
- 图表解读:正方形为 lncRNA,圆形为蛋白编码基因;红色为上调(avg_log2FC > 0),蓝色为下调(avg_log2FC < 0)。
- 分析要点:观察网络的中心节点与关键 lncRNA,以及与之相连的功能基因群;可进一步结合节点度数/介数中心性识别“枢纽”或“桥接”lncRNA,作为优先验证对象。
5.3.2 模块划分
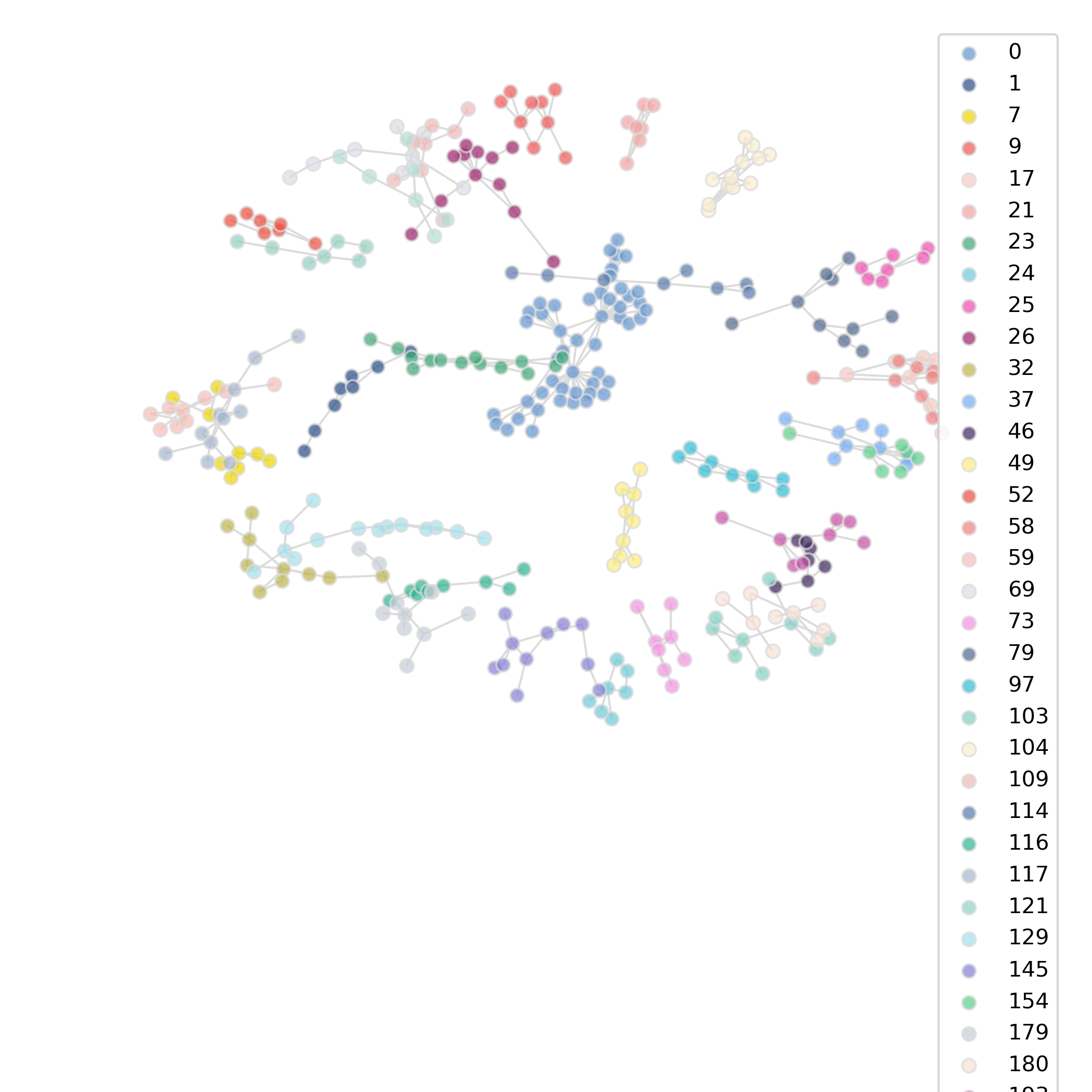
- 图表解读:不同颜色表示不同社区(模块);右上角图例数字是模块编号。
- 分析要点:使用 Louvain 算法进行共表达模块划分,过滤掉 degree 小于 7 的模块。每个模块代表在表达层面高度一致、潜在参与同一生物过程的基因集合;可对模块进行“标志基因”与“功能通路”的双重注释,以增强生物学解释的说服力。
5.4 模块功能富集 (按簇)
5.4.1 GO 富集 (示例模块)
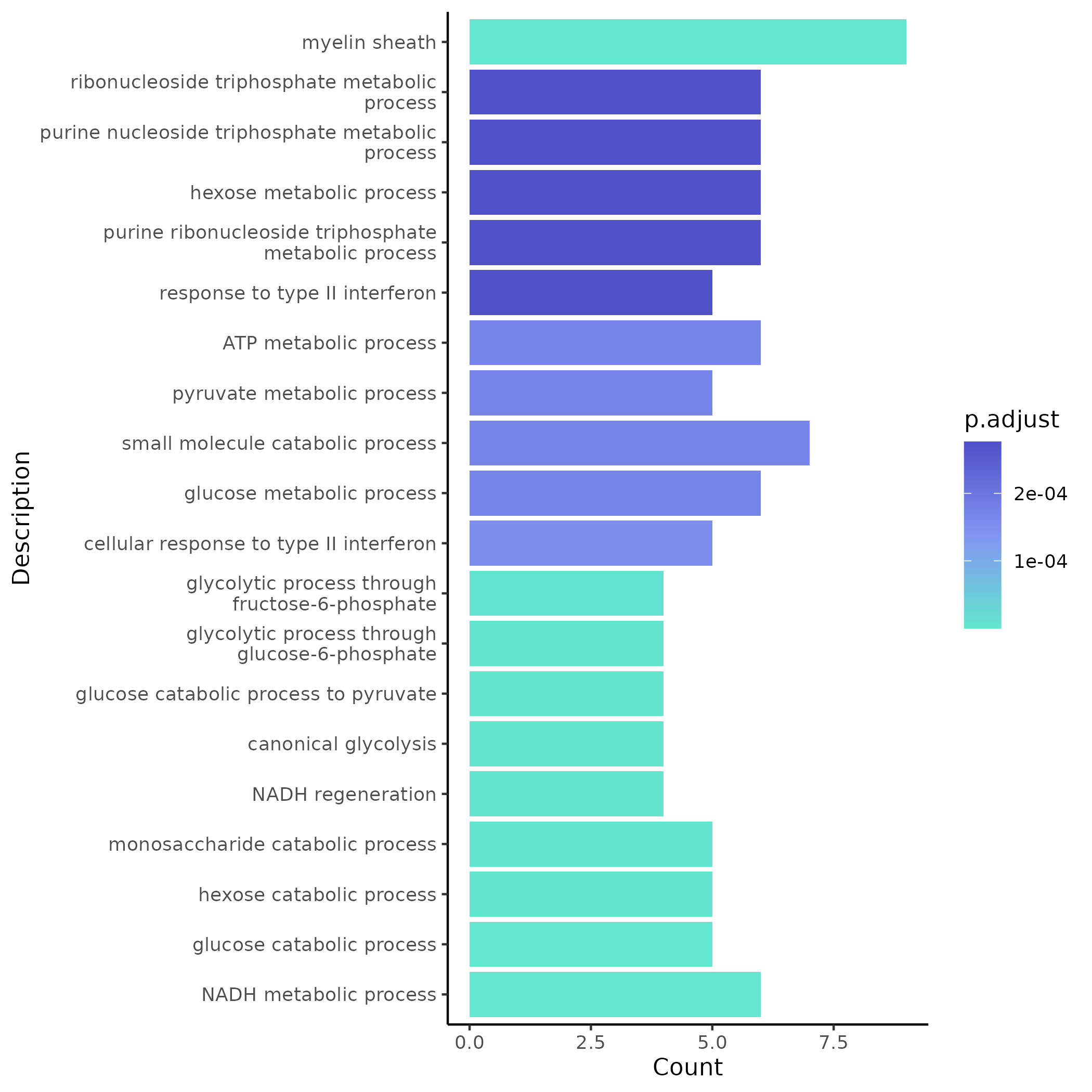
- 图表解读:x 轴为富集到该 term 的基因数,y 轴为 GO term 描述;颜色反映显著性(p.adjust),颜色越浅绿表示显著性越高。
- 分析要点:优先关注与研究主题高度相关的 GO 分支(BP/CC/MF),结合 p.adjust(显著性)与 GeneRatio(比例)综合评估;可回溯富集到的基因清单,检查是否包含核心 lncRNA 及其直接相邻的共表达基因。
5.4.2 KEGG 富集 (示例模块)
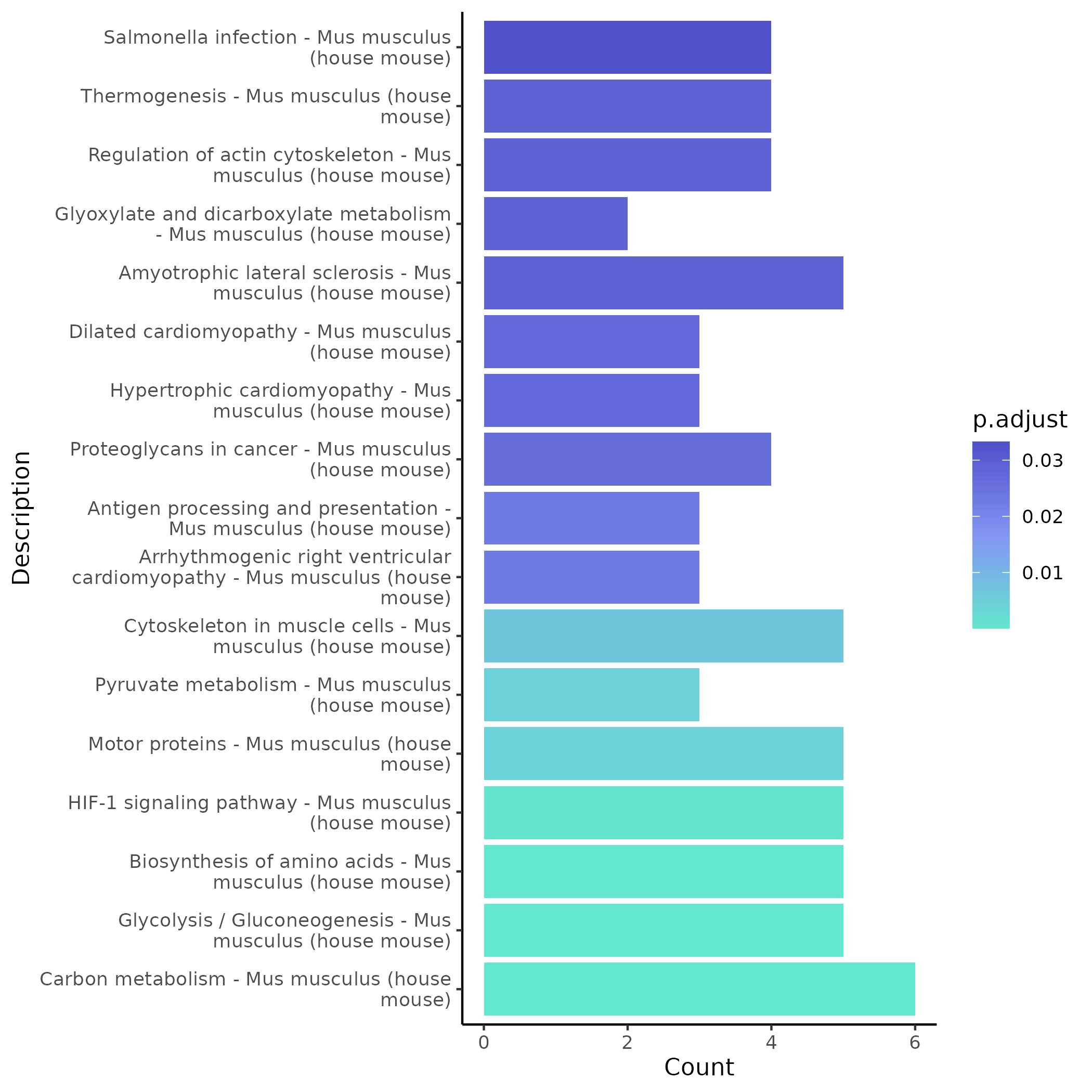
- 图表解读:横轴为富集到该通路的基因数,纵轴为 KEGG 通路名称;颜色反映显著性(p.adjust),颜色越浅绿越显著。
- 分析要点:重点关注与项目相关的经典信号通路(如免疫反应、细胞周期、凋亡、炎症、代谢与肿瘤相关通路等);建议与 GO 结果交叉验证,优先采信在多数据库均显著的通路作为结论依据。
5.5 组间比较分析
当配置了比较组时将对两组及其各簇开展差异与共表达模块分析。
5.5.1 分组 UMAP 概览
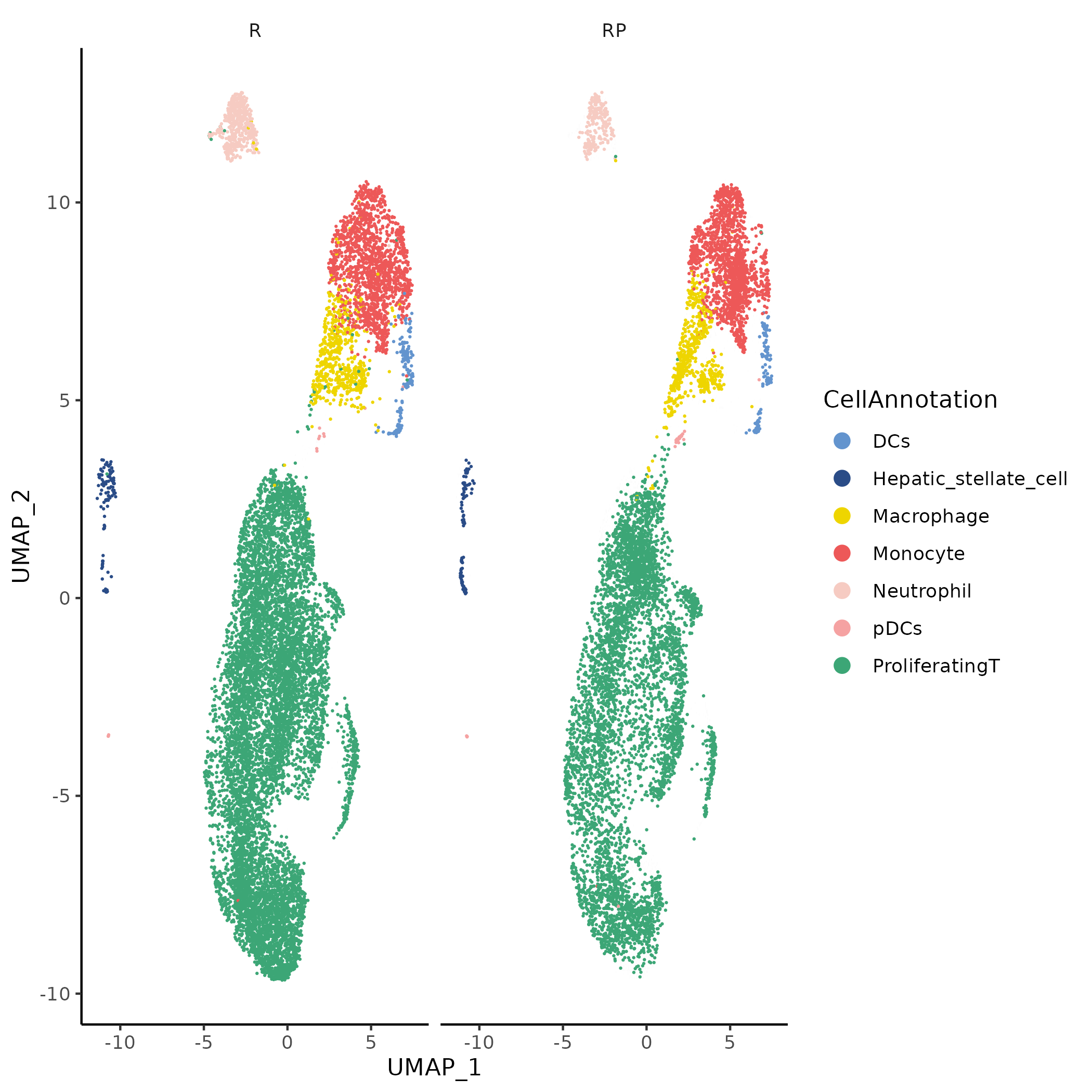
- 图表解读:按样本/组别(如
Sample/Group)拆分显示 UMAP;每个子图颜色代表细胞簇/注释标签,便于对比不同组在全局结构中的分布差异。 - 分析要点:观察处理组与对照组在细胞组成与空间位置上的变化,识别是否存在明显的细胞亚群比例差异或群落重排,为解读组间差异提供背景信息。
5.5.2 各组的 nCount/nFeature 分布
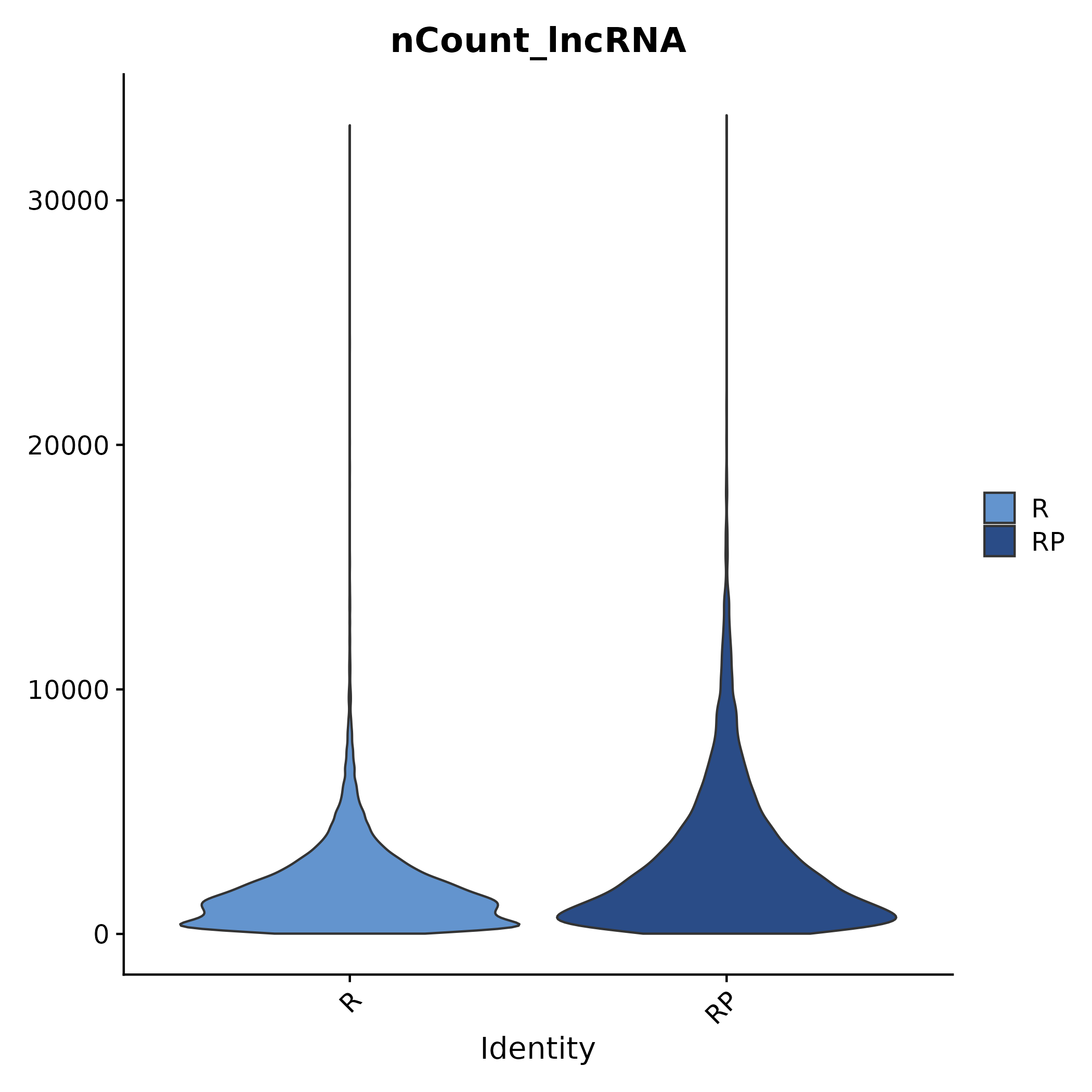
- 图表解读:展示比较组在 lncRNA 或 protein-coding RNA UMI数(nCount)与基因数(nFeature)上的分布差异,可为小提琴图/箱线图等形式。
- 分析要点:用于评估不同组之间测序深度与检测到的基因数是否均衡;若差异显著,需在差异与网络解读时谨慎,或考虑加入协变量/标准化策略。
5.5.3 差异结果可视化
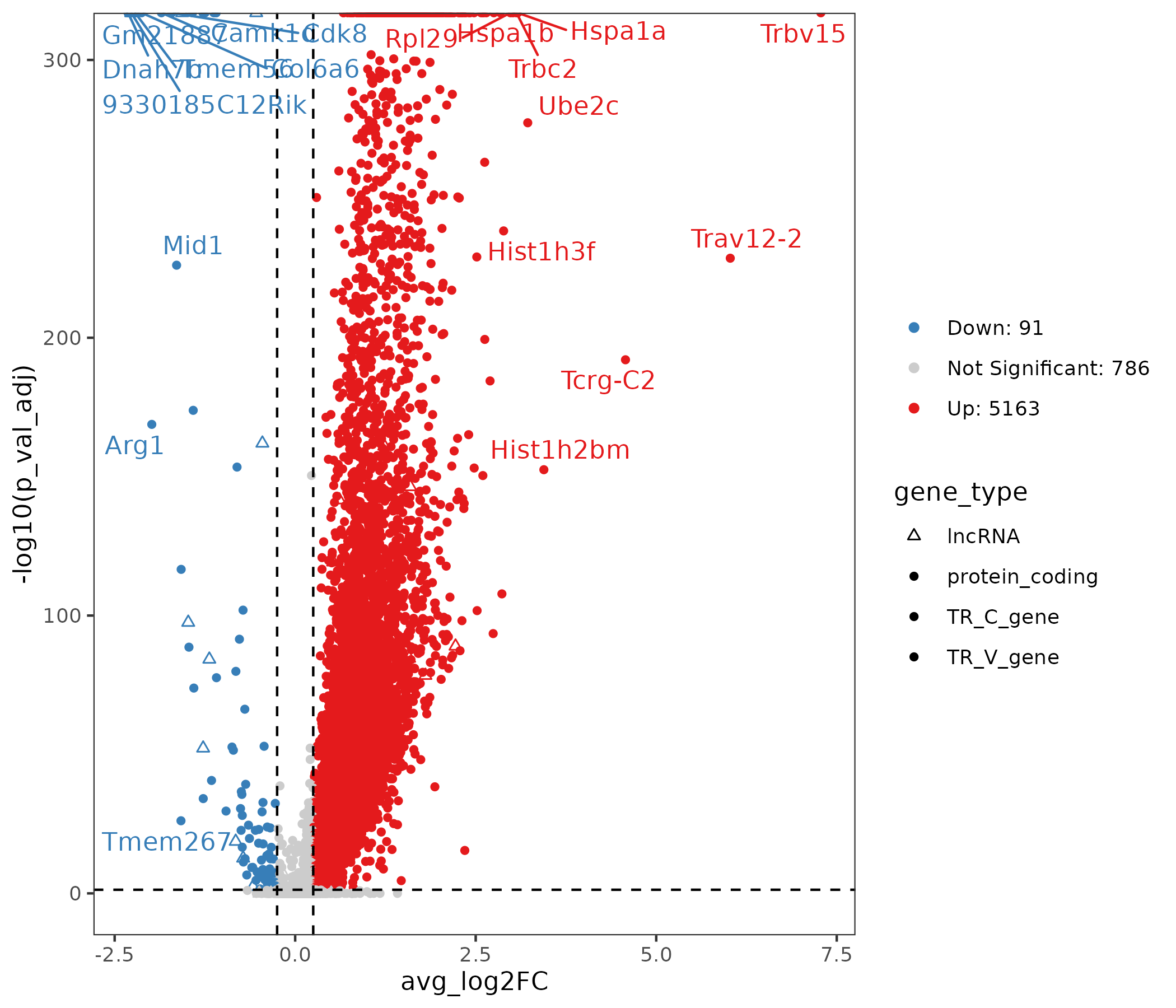
- 图表解读:横轴为效应量(log2FC),纵轴为显著性(-log10 p.adjust);点代表基因,红色代表高表达基因,蓝色代表低表达基因,灰色代表差异不显著的基因;右侧列出表达上调和下调的基因数量;形状表示不同的基因类型。
- 分析要点:关注显著且效应量大的 lncRNA 与关键蛋白编码基因;结合 Tau 与网络结果筛选稳健候选。
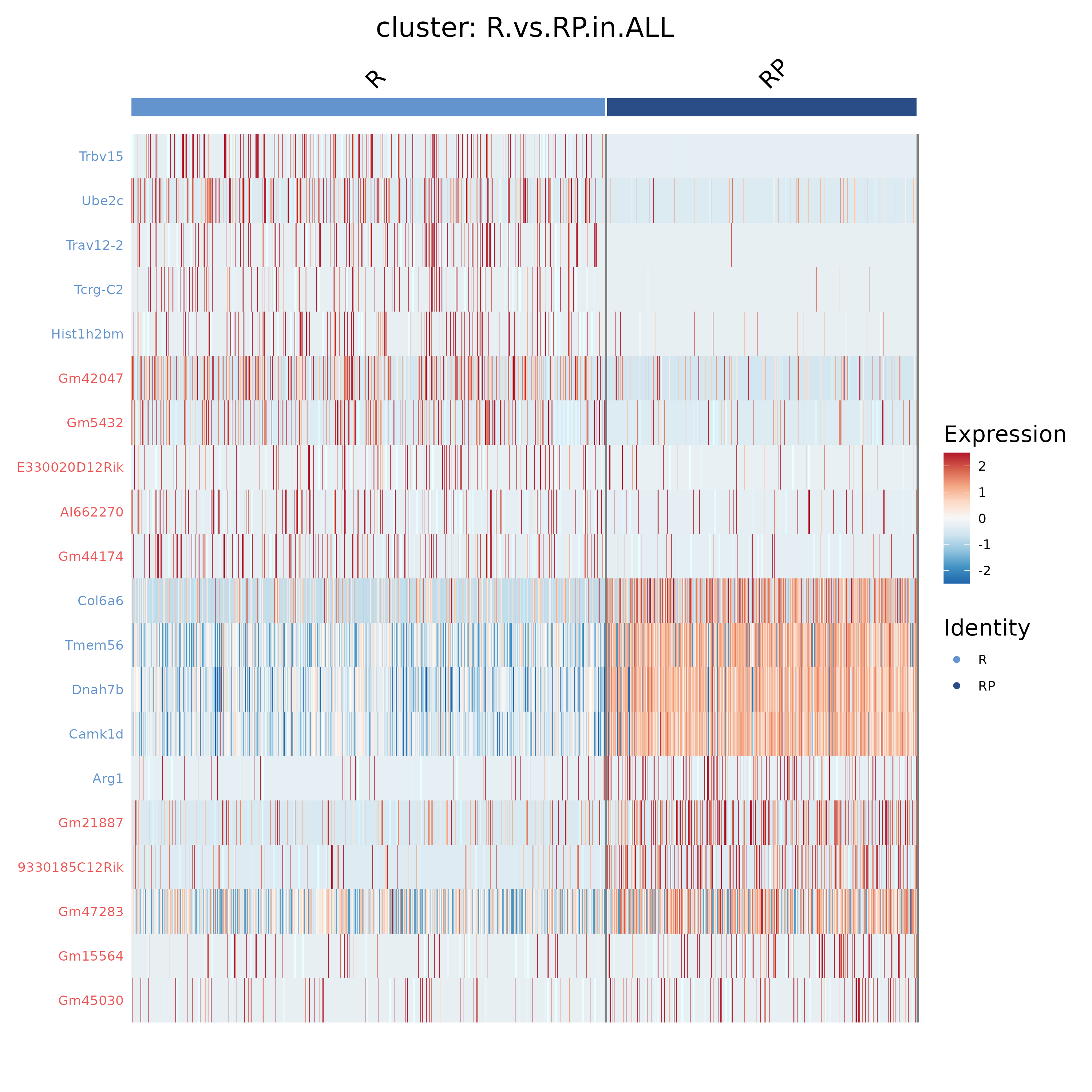
- 图表解读:行为差异基因,列为细胞;颜色表示表达强弱,颜色越红代表基因表达量越高,越蓝表示基因表达量越低。
- 分析要点:检视差异基因在不同组的整体表达模式是否清晰分离;如出现亚群特异模式,建议结合 5.5.4 的网络结果解释其功能关联。
5.5.4 比较组的共表达网络与模块
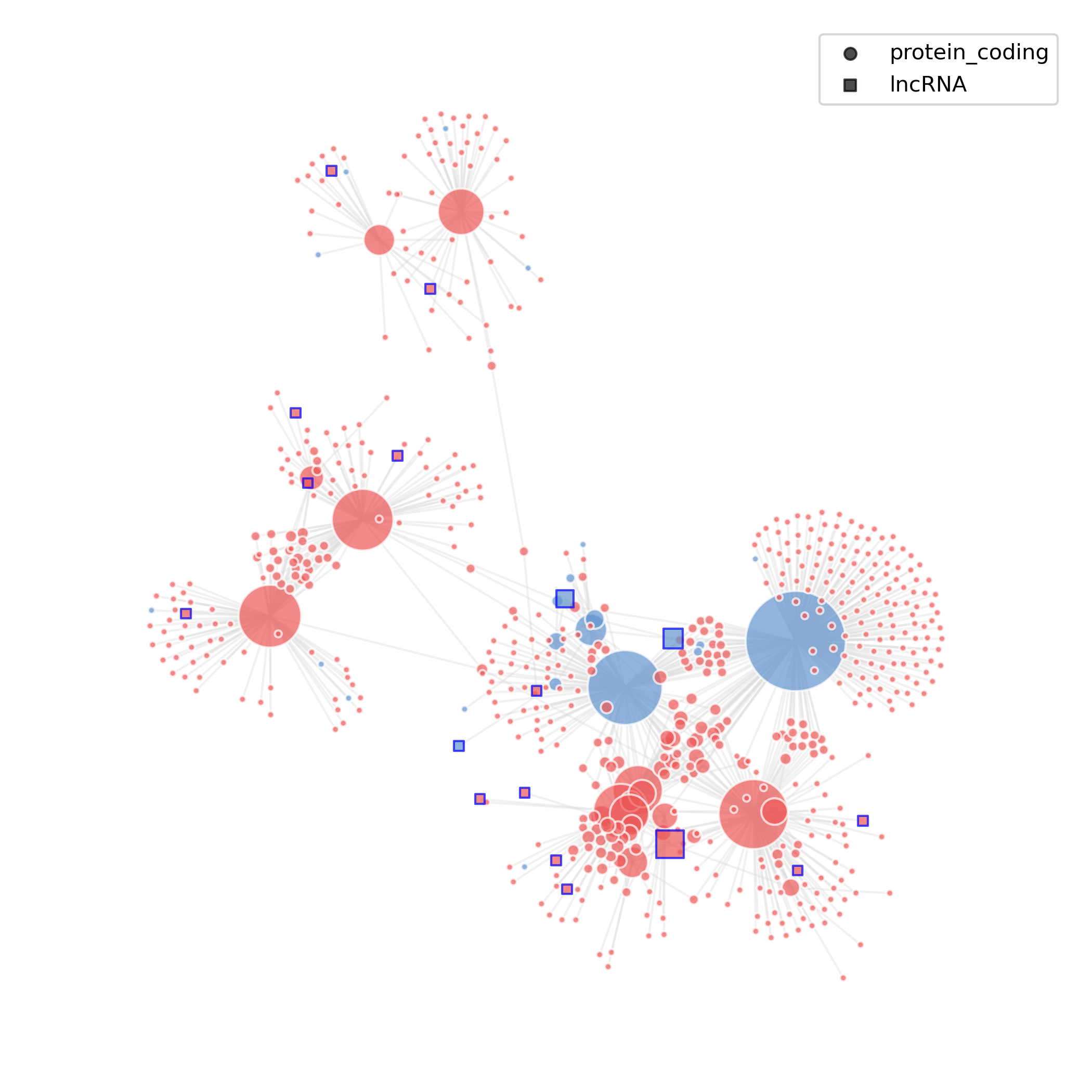
- 图表解读:节点为基因(方形为 lncRNA、圆形为蛋白编码基因),边表示高置信的共表达关系;节点颜色可代表在比较中上下调方向,红色代表高表达基因,avg_log2FC > 0,蓝色代表低表达基因,avg_log2FC < 0。
- 分析要点:识别处理组特有或显著变化的关键 lncRNA 中心节点及其邻域,推测其潜在调控作用与受影响的功能基因群。
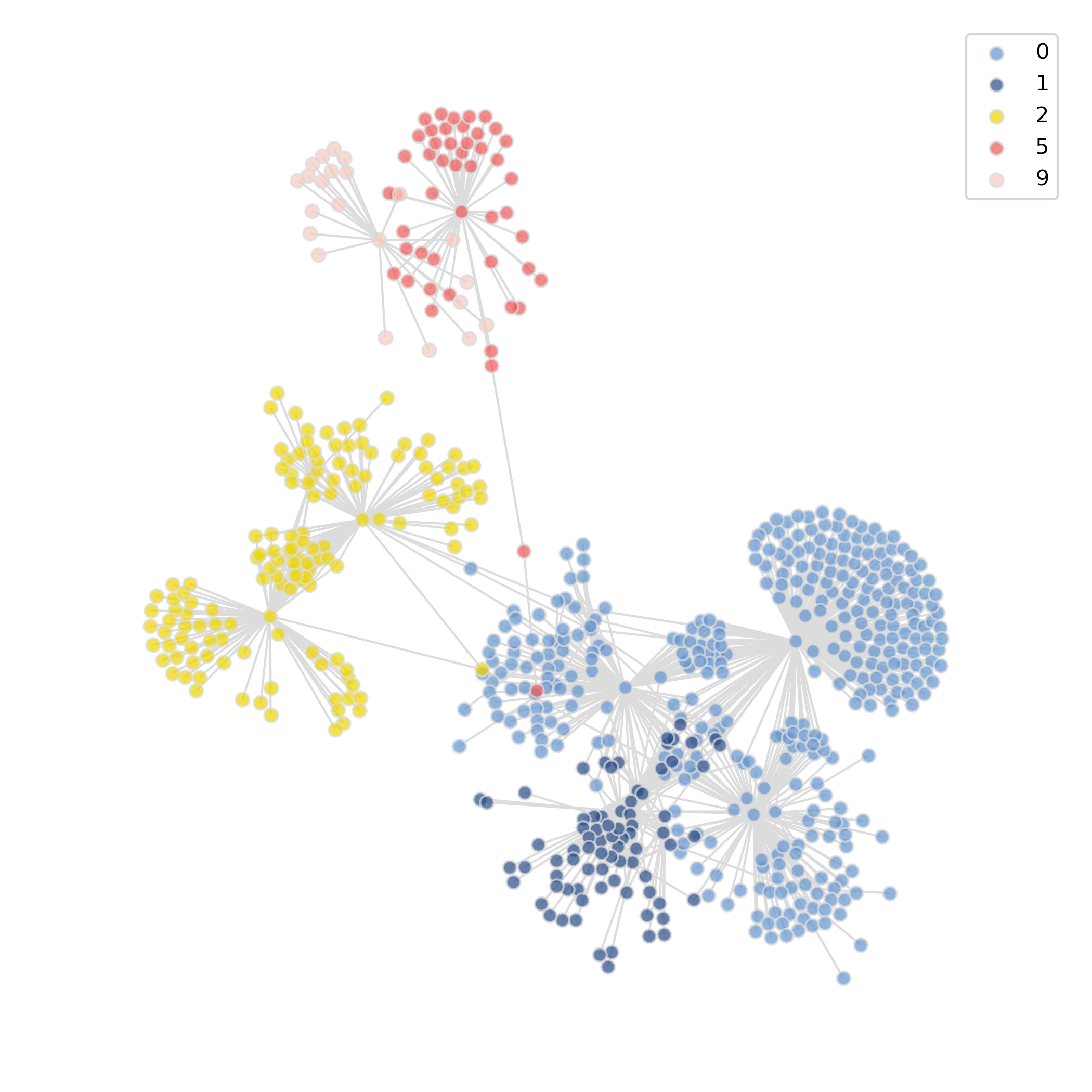
- 图表解读:不同颜色代表不同社区(功能模块),社区内基因在表达上高度一致,可能参与同一生物过程。
- 分析要点:比较不同组的模块组成与规模,识别新出现或显著变化的模块,以锁定处理驱动的网络重构信号。
5.5.5 比较组的模块富集
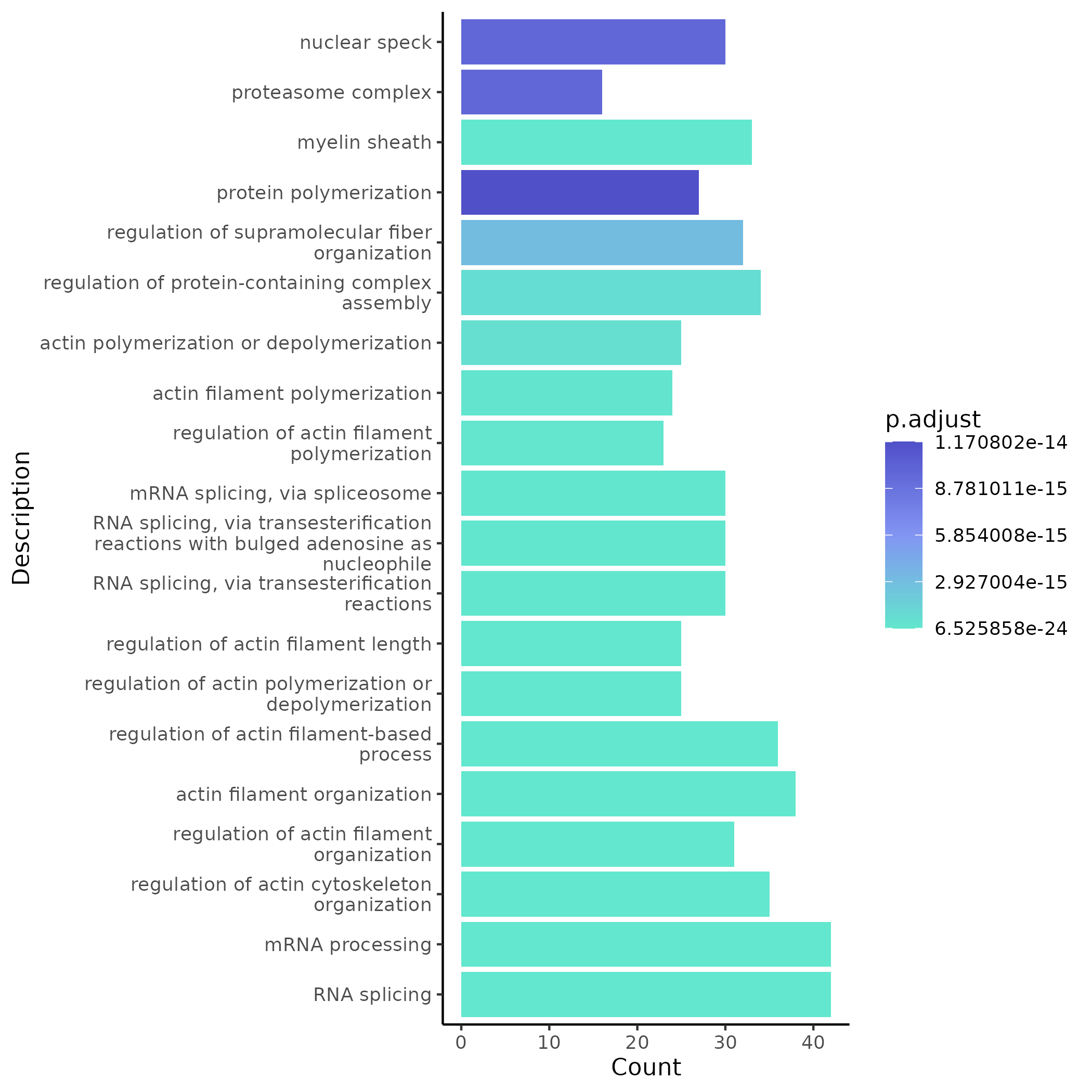
- 图表解读:展示比较场景下显著模块的 GO 富集条形图,指示各模块潜在的生物学功能。
- 分析要点:定位仅在处理组显著的 GO term,作为处理引发功能改变的直接证据;结合网络中心基因与 lncRNA 进行生物学解释。
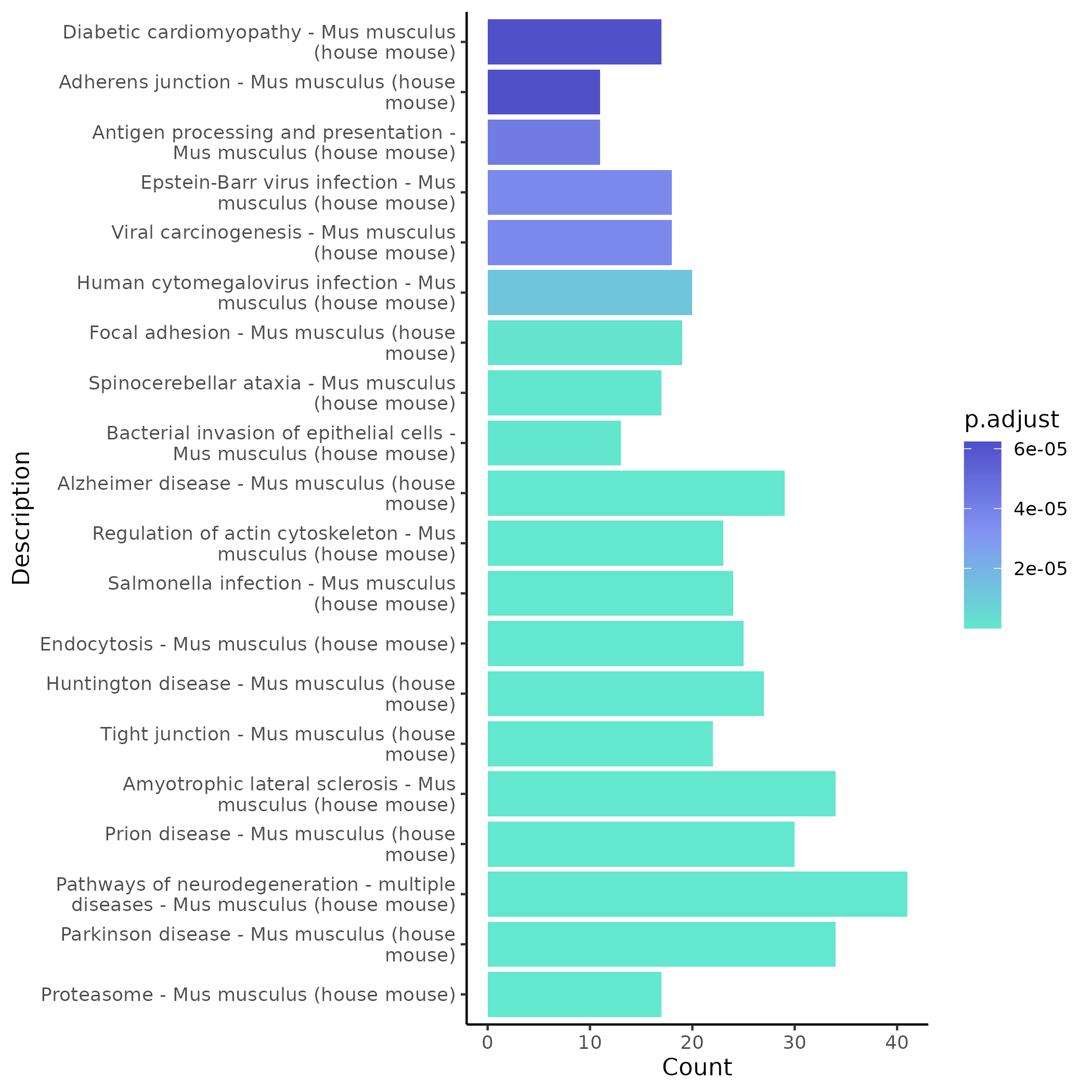
- 图表解读:展示比较场景下显著模块的 KEGG 富集条形图。
- 分析要点:重点关注与课题相关的关键信号通路(如免疫/炎症/代谢等),并与 GO 结果交叉验证,提升结论的稳健性。
TIP
比较分析可帮助识别条件/处理驱动的 lncRNA-蛋白编码基因关系改变,从网络层面揭示潜在机制差异。
5.6 结果文件列表
| 目录/文件 | 内容说明 |
|---|---|
01_lncRNA_info/ | lncRNA 表达概览相关图表与表格(如 cluster_umap.png、lnc_fraction.png、nCount_lncRNA.png、top10_lncRNA_genes.png、Tau 评分与 top4 基因可视化等)。 |
02_cor_and_get_matrix/ | 按簇计算的基因相关性与矩阵文件(如相关系数/重要性矩阵、节点/边表等),用于后续网络构建与阈值筛选。 |
03_module_plots/<Cluster>/ | 按细胞簇构建的网络与模块图(<Cluster>_network.png、cluster_<Cluster>_network_community.png),以及 GO/ 与 KEGG/ 子目录中的富集结果图表。 |
04_compare/<Pair>/ | 按比较组对(如 R.vs.RP/)的差异可视化:umap_split_by_Sample.png、nCount_lncRNA.png、*.volcano.png、*.heatmap.png 等。 |
05_compare_cor_and_get_matrix/ | 按比较场景计算的基因相关性与矩阵文件(比较版的节点/边/相关性结果),用于构建比较组网络与模块。 |
06_compare_module_plots/<Pair>/ | 按比较组对的网络与模块图,以及对应的 GO/、KEGG/ 富集结果。 |
注意事项
1. novel-lnc 步骤耗时且依赖数据质量:如仅做已知 lncRNA 共表达,建议先关闭该选项以加快流程。
2. 元数据命名规范:建议全英文,避免中文与特殊字符;不同样本/批次的命名需统一。
3. 比较策略建议:同时进行“ALL”层面与“簇内”层面的对比,以捕捉广义与细粒度差异。
常见问题解答 (FAQ)
Q1:开启“识别 novel-lnc”需要哪些前提?
A:需要提供每个样本的 bam_path;该步骤耗时较长,且对比对质量、参考注释的完备性敏感。
Q2:为什么某些簇没有得到模块或富集结果?
A:常见原因是差异基因过少或网络过稀被过滤(如 degree < 7)。
Q3:网络过于复杂难以解读怎么办?
A:优先聚焦中心度高的 lncRNA 及其相邻模块进行分层解读。
Q4:如何理解 Tau 特异性评分?
A:Tau 越接近 1 表示在少数簇中高度特异;越接近 0 表示在多簇中普遍表达。可结合差异与网络位置综合评估。
Q5:结果能用于动物/人跨物种比较吗?
A:本模块支持 human/mouse;跨物种比较需额外的基因同源映射步骤,非本模块内置能力。
Q6:比较组的“拆分因子/处理组/对照组”如何选择?
A:建议优先采用能代表生物学处理与对照的标签(如 Group/Sample),并保证两组在细胞数量与质控标准上尽量平衡。
参考资料
[1] SANTUS L, SOPENA-RIOS M, GARCÍA-PÉREZ R, et al. Single-cell profiling of lncRNA expression during Ebola virus infection in rhesus macaques[J]. Nat Commun, 2023, 14: 3866.
[2] VAN DE SANDE B, FLERIN C, DAVIE K, et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis[J]. Nature Protocols, 2020, 15: 2247–2276.
[3] HAO Y, HAO S, ANDERSEN-NISSEN E, et al. Integrated analysis of multimodal single-cell data[J]. Cell, 2021, 184: 3573–3587.
[4] KIM D, PAGGI J M, PARK C, et al. Graph-based genome alignment and genotyping with HISAT2[J]. Nat Methods, 2019, 16: 887–938.
[5] DOBIN A, DAVIS C A, SCHLESINGER F, et al. STAR: ultrafast universal RNA-seq aligner[J]. Bioinformatics, 2013, 29: 15–21.