SeekSoul Tools v1.2.2
SeekSoul Tools is a software developed by SEEKGENE for processing single-cell transcriptome data. Currently, the software contains four modules.
1.rna module;This module is used for identifying cell barcode, genome alignment and gene quantification to obtain a feature-barcode matrix that can be used for downstream analsis, followed by cell clustering and differential analysis. This module not only support data from SeekOne™ transcriptome kits, it also supports a variety of customized structure designs.
2.fast module:This module is specifically designed for data produced by the SeekOne™ DD Single Cell Full-length RNA Sequence Transcriptome-seq Kit, which is used for barcode extraction, paired-end read alignment, quantification, and unique metrics analysis for full-length transcriptome data.
3.vdj module: This module is specifically designed for data produced by the SeekOne™ DD Single Cell Immune Profiling Kit, which is used for assembling, filtering, and annotating immune receptors.
4.utils module: The module contains other small tools.
Download
SeekSoul Tools v1.2.2
Download-SeekSoul Tools - md5: af52b5d936b60c740b239301f2026aba
wget
mkdir seeksoultools.1.2.2
cd seeksoultools.1.2.2
wget -c -O seeksoultools.1.2.2.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.2.tar.gz"curl
mkdir seeksoultools.1.2.2
cd seeksoultools.1.2.2
curl -C - -o seeksoultools.1.2.2.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.2.tar.gz"Installation Guide
IMPORTANT
Before installation, please ensure that the conda environment is properly configured to avoid conflicts with other bioinformatics software.
Installation:
# decompress
tar zxf seeksoultools.1.2.2.tar.gz
# export path
export PATH=`pwd`:$PATH
echo "export PATH=$(pwd):\$PATH" >> ~/.bashrc
# Initialization and installation verification
./seeksoultools --versionTIP
It is recommended to add PATH to ~/.bashrc for easy command-line access.
Verify installation:
seeksoultools --versionNOTE
If you encounter "command not found" or dependency errors, please check the conda environment activation and PATH settings.
Tutorials
rna module
Data preparation
Download sample datasets
sample datasets - md5: 3d15fcfdefc0722735d726f40ec4e324(Species: Homo sapiens.)
wget
wget -c -O demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tarcurl
curl -C - -o demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tarDownload and build reference genome
Download-human-reference-GRCh38 - md5: 5473213ae62ebf35326a85c8fba6cc42
Download-mouse-reference-mm10 - md5: 5c7c63701ffd7bb5e6b2b9c2b650e3c2
wget
wget -c -O GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gzcurl
curl -C - -o GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gzThe assembly of the reference genome refers to How to build reference genome?
CAUTION
When downloading and decompressing the reference genome, please ensure sufficient disk space to avoid file corruption that could lead to subsequent analysis failure.
Run SeekSoulTools
Run tests
Example 1: Basic usage
Set up the necessary configuration files for the analysis, including the paths to the sample data, the chemistry versions, the genome index, the gene annotation file, etc. Run the SeekSoulTools using the following command:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-intronsExample 2: Specify a different version of STAR for analysis.
To use a specific version of STAR for analysis while ensuring compatibility with the –genomeDir generated by that version, you can run the SeekSoulTools using the following command, specifying the path to the desired version of STAR:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-introns \
--star_path /path/to/cellranger-5.0.0/lib/bin/STARNOTE
The --star_path parameter must strictly correspond to the reference genome index version, otherwise alignment will fail.
Example 3: A sample has multiple sets of fastq files
If a sample has multiple sets of FASTQ data, you can provide the paths to all the FASTQ files associated with that sample when running the SeekSoulTools. Here's an example command:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq1 /path/to/demo_dd_S39_L002_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L002_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-intronsExample 4: Customize the structure of R1
To customize the structure of the Read 1 (R1) FASTQ files, here's an example command:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--barcode /path/to/utils/CLS1.txt \
--barcode /path/to/utils/CLS2.txt \
--barcode /path/to/utils/CLS3.txt \
--linker /path/to/utils/Linker1.txt \
--linker /path/to/utils/Linker2.txt \
--structure B9L12B9L13B9U8 \
--core 4 \
--include-introns- The structure of read1 is represented by
B9L12B9L13B9U8, which means it consists of three sections of cell barcode, each with 9 bases, and a UMI section with 8 bases. The linker section between the cell barcode and UMI consists of two parts, with the first part being 12 bases and the second part being 13 bases - Use
--barcodeto specify the three sections of barcodes sequentially, and use--linkerto specify the two sections of linkers sequentially.
TIP
When customizing the structure, it is recommended to test with a small sample first to ensure barcode/linker/UMI extraction is correct.
Parameter descriptions
| Parameters | Descriptions |
|---|---|
| --fq1 | Paths to R1 fastq files. |
| --fq2 | Paths to R2 fastq files. |
| --samplename | Sample name. A directory will be created named after the sample name in the outdir directory. Only digits, letters, and underscores are supported. |
| --outdir | Output directory. Default: ./ |
| --genomeDir | The path of the reference genome generated by STAR. The version needs to be consistent with the STAR used by SeekSoulTools. |
| --gtf | Path to the GTF file for the corresponding species. |
| --core | Number of threads used for the analysis. |
| --chemistry | Reagent type, with each type corresponding to a combination of --shift,--pattern, --structure, --barcode, and--sc5p. Available options: DDV2, DD5V1, MM, MM-D. DDV2 corresponds to the SeekOne™ DD Single Cell 3' Transcriptome-seq Kit. DD5V1 corresponds to the SeekOne™ DD Single Cell 5' Transcriptome-seq Kit. MM corresponds to the SeekOne™ MM Single Cell Transcriptome Kit. MM-D corresponds to the SeekOne™ MM Large-well Single Cell Transcriptome-seq Kit. |
| --skip_misB | If enabled, no base mismatch is allowed for barcode. Default is 1. |
| --skip_misL | If enabled, no base mismatch is allowed for linker. Default is 1. |
| --skip_multi | If enabled, discard reads that can be corrected to multiple white-listed barcodes. Barcodes are corrected to the barcode with the highest frequency by default. |
| --expectNum | Estimated number of captured cells. |
| --forceCell | When number of cells obtained from analysis is abnormal, add this parameter with expected value N. SeekSoulTools will select the top N cells based on UMI from high to low. |
| --include-introns | When disabled, only exon reads are used for quantification. When enabled, intron reads are also used for quantification. |
| --star_path | Path to another version of STAR for alignment. The version must be compatible with the --genomeDir version. The default --star_path is the STAR in the environment. |
WARNING
Incorrect parameter settings (such as chemistry, genomeDir, gtf, etc.) will directly lead to analysis failure or abnormal results. Please be sure to verify parameter meanings and input paths.
Output descriptions
Here's the output directory structure: each line represents a file or folder, indicated by "├──", and the numbers indicate three important output files.
./
├── demo_report.html 1
├── demo_summary.csv 2
├── demo_summary.json
├── step1
│ └──demo_2.fq.gz
├── step2
│ ├── featureCounts
│ │ └── demo_SortedByName.bam
│ └── STAR
│ ├── demo_Log.final.out
│ └── demo_SortedByCoordinate.bam
├── step3
│ ├── filtered_feature_bc_matrix 3
│ └── raw_feature_bc_matrix
└──step4
├── FeatureScatter.png
├── FindAllMarkers.xls
├── mito_quantile.xls
├── nCount_quantile.xls
├── nFeature_quantile.xls
├── resolution.xls
├── top10_heatmap.png
├── tsne.png
├── tsne_umi.png
├── tsne_umi.xls
├── umap.png
└── VlnPlot.pngNOTE
If any files are missing from the output directory, please check the log file for error messages and ensure all necessary input files and parameters are set correctly.
- Final report in html
- Quality control information in csv
- Filtered feature-barcode matrix
Algorithms Overview
step1: barcode/UMI extraction
SeekSoulTools is able to extract the barcode and UMI sequences based on different Read1 structures. The pipeline processes the barcodes and filters Read1 and its corresponding Read2, and an updated fastq file is generated at the end of step 1.
Structure design and description
Barcode and UMI descriptions: Describing the basic structure of Read1 using letters and numbers, where the letters represent the meaning of the nucleotides and the numbers represent the length of the nucleotides.
B: barcode bases L: linker bases U: UMI bases X: other arbitrary bases used as placeholders
Taking the following two Read1 structures as examples: B8L8B8L10B8U8:

B17U12:

Anchor-based misalignment design: In MM design, to increase the base balance of the linker portion during sequencing,1-4 bp shifted bases, called anchors, were added. The anchor determines the starting position of the barcode.

Workflow
Anchor determination:
For data with misaligned Read1 (produced by MM reagents), SeekSoulTools attempts to find the anchor sequence within the first 7 bases of the Read1 sequence to determine the start position of subsequent barcodes. If the anchor sequence is not found, the corresponding Read1 and R2 are considered invalid reads.
Barcode and linker correction:
After determining the starting position of the barcode, the corresponding sequence is extracted based on the structural design. When the extracted barcode sequence is found in the whitelist, it is considered a valid barcode and the read count with valid barcodes is recorded. When the barcode is not found in the whitelist, it is considered an invalid barcode.
During sequencing process, there is a certain probability that sequencing error occurs. With the presense of a whitelist, SeekSoulTools are able to do barcode correction. When correction is enabled, if sequences that are one base differ (one humming distance apart) from the invalid barcode appear in the whitelist, we will consider the following circumstances:
- If there is one and only sequence exists in the whitelist, we will correct the invalid barcode to the barcode in the whitelist.
- If multiple sequences exist in the whitelist, we will correct the invalid barcode to the sequence with the highest read support.
The logic of linker correction is the same as that of barcode correction.
Adaptors and polyA sequence trimming
In transcriptome, polyA tail and Adapter sequences introduced during library preparation, may appear at the end of Read2.We remove these contaminating sequences and make sure the trimmed Read2 length is greater than the minimum length for accurate alignment afterward. If the trimmed Read2 length is less than the minimum length, we consider the read to be invalid.
After the processing procedure described above, the data composition is shown in the following figure:
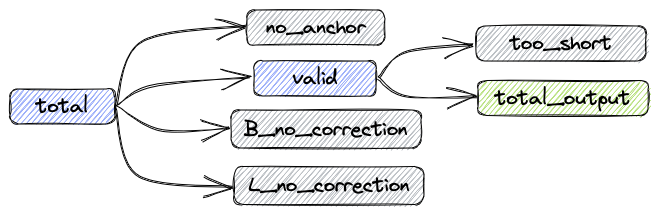
- total: Total reads
- valid: Number of reads without correction and number of successfully corrected reads
- B_corrected: Number of successfully corrected reads
- B_no_correction: Number of reads with incorrect barcodes
- L_no_correction: Number of reads with incorrect linkers
- no_anchor:Number of reads without anchors
- trimmed: Number of reads that have been trimmed
- too_short: Number of reads that are shorter than 60bps after trimming
The relationships between the metrics are as follows:
total = valid + no_anchor + B_no_correction + L_no_correction
Number of reads in the updated FASTQ file: total_output = valid - too_short
step2: Alignment
Sequence Alignment
- SeekSoulTools uses an aligner software called STAR to perform splicing-aware alignment of reads to the genome.
- After the alignment procedure has mapped the reads to the genome, SeekSoulTools uses another software called QualiMap along with a gene annotation transcript file GTF to bucket the reads into exonic, intronic, and intergenic regions.
- Using featureCounts, the reads aligned to the genome were annotated to genes, with different rules such as strand specificity and features used for annotation. When using exon for annotation, a read is annotated to the corresponding gene if over 50% of its bases are aligned to the exon. When using transcripts for annotation, a read is annotated to the corresponding gene if over 50% of its bases are aligned to the transcript.
After the processing procedure described above, the metrics is shown below:
- Reads Mapped to Genome: Fraction of reads that aligned to the genome (including both uniquely mapped reads and multi-mapped reads)
- Reads Mapped Confidently to Genome: Fraction of reads that uniquely mapped to the genome
- Reads Mapped to Intergenic Regions:Fraction of reads that mapped to intergenic regions
- Reads Mapped to Intronic Regions:Fraction of reads that mapped to intronic regions
- Reads Mapped to Exonic Regions:Fraction of reads that mapped to exonic regions
step3: Counting
UMI counting
SeekSoulTools extracts group of reads that shared the same barcode from output BAM file and counts the number of UMIs annotated to genes and the number of reads corresponding to each UMI.
- Filter out reads whose UMIs consist of repetitive bases. For example, TTTTTTTT.
- If a read has multiple annotations and only one gene annotation is from exon, it is considered a valid read, and all others are filtered.
UMI correction
UMIs may also have sequencing errors during sequencing process. By default, SeekSoulTools uses the adjacency from UMI-tools to correct UMIs.
 Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Cell calling
In a cell population, we assume that the mRNA content of cells does not differ significantly. If the mRNA content of a barcode is very low, we consider that barcode might be 'empty', and the mRNA comes from the background. SeekSoulTools uses the following steps to determine whether a barcode has a cell:
- Sort all barcodes in descending order based on their corresponding UMI counts.
- The threshold is calculated by dividing the number of UMIs for the barcode at the 1% position of the estimated cells by 10.
- Barcodes with UMIs greater than the threshold are considered to be cells.
- If the UMI of a barcode is less than the threshold but greater than 300, DropletUtils is used for further analysis. The DropletUtils method first assumes that barcodes with UMI counts below 100 are empty droplets. It then uses the total UMI count for each gene in each droplet as the expected UMI count for that gene in the background RNA expression profile. Next, it performs a statistical test on the UMI counts of each barcode, identifying those with significant differences as cells.
- Those do not meet the above criteria are considered as background.
After the processing procedure described above, the metrics is shown below:
- Estimated Number of Cells: Total number of cells by cell calling
- Fraction Reads in Cells: Fraction of reads after cell calling among all reads used for counting
- Mean Reads per Cell: Average number of reads per cell, Total number of reads/Number of cells after cell calling
- Median Genes per Cell: Median gene count in barcodes after cell calling
- Median UMI Counts per Cell: Median UMI count in barcodes after cell calling
- Total Genes Detected: Number of genes detected in all cells
- Sequencing Saturation: 1 - Total UMI count/Total read count
step4: Downstream analysis
SeekSoulTools perform downstream analysis when we have gene expression matrix from step3.
Seurat analysis
SeekSoulTools use Seurat to calculate the mitochondrial content, number of genes, and UMIs of each cell. After that, the gene expression matrix is normalized, and a subset of features that exhibit high cell-to-cell variation in the dataset is identified. Linear dimensional reduction using PCA is then performed, and the result is passed to t-SNE and UMAP for visualization. A graph-based clustering procedure is then followed, and cells are partitioned into different clusters. Finally, SeekSoulTools finds markers that define clusters via differential expression.
fast module referencefast module
vdj module referencevdj module
utils module
addtag
Modify BAM files to include barcode and UMI tags.
Run tests
Set up necessary configuration files for analysis, including the sample's BAM file and the umi.xls file in the step3 directory. Run the SeekSoulTools using the following
seeksoultools utils addtag \
--inbam step2/featureCounts/Samplename_SortedByName.bam \
--umifile step3/umi.xls \
--outbam Samplename_addtag.bamParameter descriptions
| Parameters | Descriptions |
|---|---|
| --inbam | sample's BAM file in the step2/featureCount directory. |
| --outbam | The path of the BAM file with added tags. |
| --umifile | he path of the umi.xls file in the step3 directory. |
FAQ
How to build reference genome?
The assembly of the reference genome refers to How to build reference genome?
Release Notes
v1.2.2
- Add vdj analysis module
- Support for FFPE samples in the fast module
- Fix the statistical error in gene median calculation caused by missing lncRNA type in the GTF file.
- Resolve the issue of missing Ensembl IDs for differentially expressed genes due to gene name duplication.
- Other improvements to enhance usability.
v1.2.1
- Update the style of the report
- Add trimming for SP1, SP2, TSO, and adapters
- Add tool for adding tags to BAM files
- Enhance support for non-standard GTF files
- Enhance the FAST module to include support for species other than human and mouse
v1.2.0
- Add output of read1 fastq file after removing barcode and UMI sequences
- Add fast analysis module
- Apply UMI-tools correction method
- Update rules for handling annotations of multiple genes in reads: When there is a unique exon annotation, consider the read as valid
v1.0.0
- Initial release