Analysis Guide
Time: 3 min
Words: 519 words
Updated: 2026-05-12
Reads: 0 times
Activate the Environment
bash
conda activate seeksoulmethylRun the Dual-omics Workflow with the Shell Script
bash
bash sc_methy_workflow.sh \
/path/to/expression_R1.fastq.gz \
/path/to/expression_R2.fastq.gz \
/path/to/methy_R1.fastq.gz \
/path/to/methy_R2.fastq.gz \
--sample WTJW880 \
--outdir /path/to/results \
--database_dir /path/to/human-reference-GRCh38 \
--chemistry DD-MET3 \
--core 64 \
--filter_ch 2For samples with multiple datasets, provide comma-separated FASTQ lists in matching order.
System Requirements for Shell Mode
Recommended resources for sc_methy_workflow.sh:
- CPU: 64 cores
- Memory: 128 GB RAM
- Storage: at least 500 GB free space
- Operating system: Linux, preferably Ubuntu 18.04+ or CentOS 7+
Run the Workflow with Nextflow
Nextflow is the recommended method for batch processing, report generation, and workflow management.
Install Nextflow
bash
conda install -n seeksoulmethyl -c bioconda nextflowRun the Main Workflow
bash
nextflow run -bg SeekSoulMethyl/nf/main.nf \
--outdir /path/to/results/ \
--samplesheet samplelist.csv \
-w /path/to/results/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/human-reference-GRCh38/ \
--split_fastq 1 \
--filter_ch 2 \
--chemistry DD-MET3 > methy.log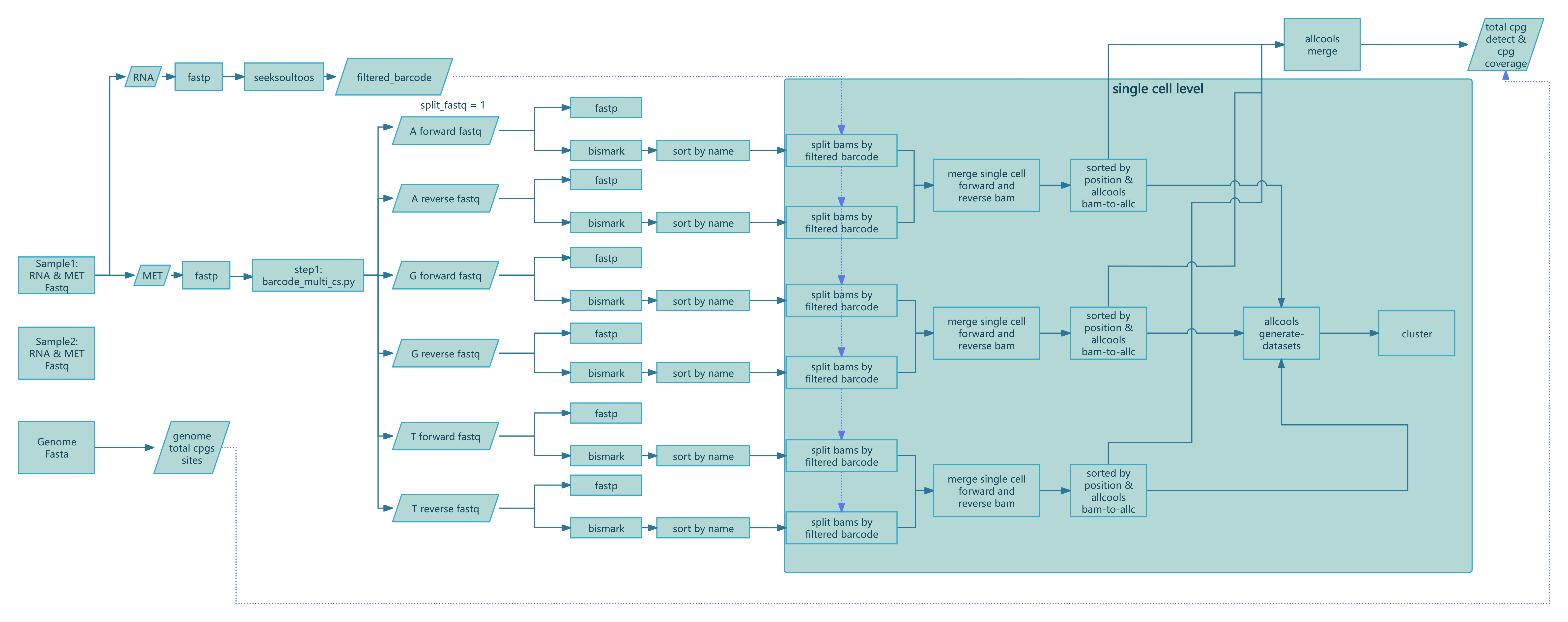
Figure 1: SeekSoul™ Methyl Tools Nextflow pipeline workflow
Supported Nextflow Workflows
The v2.1.2 code base supports multiple workflows through --workflow:
rna_met: transcriptome plus methylation integrated analysismethy_only: methylation-only workflowforce_cell: recompute or update methylation results based on previous outputs
Methylation-only Workflow
bash
nextflow run SeekSoulMethyl/nf/main.nf \
--workflow methy_only \
--outdir /path/to/results \
--samplesheet samplelist.csv \
-w /path/to/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/reference \
--split_fastq 4 \
--filter_ch 2 \
--chemistry DD-MET3Notes on nextflow.config
The file nf/nextflow.config must be adjusted for your infrastructure. Focus on these items:
process.executor: chooselocal,slurm,pbs,k8s,awsbatch, or another supported executorprocess.cpus,process.memory,process.time: default resource limitsworkDir: working directory with enough writable storageconda.enabled,docker.enabled, orsingularity.enabled: select the environment strategy your platform supports
Typical profiles include local execution, Slurm clusters, and Kubernetes environments.
Key Runtime Parameters
--database_dir: reference genome database directory--chemistry: chooseDD-MET3orDD-MET5--split_fastq: split methylation FASTQ files by the firstnbarcode bases to increase parallelism--filter_ch: remove read pairs with more thannCH methylation sites; set to0to disable-resume: restart a failed or interrupted Nextflow run with the same work directory
FAQ
- Samplesheet parsing error: confirm the first column is exactly
sample_idand use absolute file paths. - Missing
${sample}.mcds: check whether per-cell*_allc.gzfiles were generated and confirm chromosome-size resources are valid. - Bismark alignment stalls or fails: confirm the Bismark reference is present under
--database_dir/fasta/and visible to the execution environment. - Repeated runs: keep the same
-wworking directory and add-resume.