分析指南
时长: 3 分钟
字数: 743 字
更新: 2026-05-12
阅读: 0 次
激活环境
bash
conda activate seeksoulmethyl使用 Shell 脚本运行双组学工作流
bash
bash sc_methy_workflow.sh \
/path/to/expression_R1.fastq.gz \
/path/to/expression_R2.fastq.gz \
/path/to/methy_R1.fastq.gz \
/path/to/methy_R2.fastq.gz \
--sample WTJW880 \
--outdir /path/to/results \
--database_dir /path/to/human-reference-GRCh38 \
--chemistry DD-MET3 \
--core 64 \
--filter_ch 2对于包含多个数据集的样本,请按匹配顺序提供逗号分隔的 FASTQ 列表。
Shell 模式的系统要求
运行 sc_methy_workflow.sh 的推荐资源:
- CPU:64 核心
- 内存:128 GB RAM
- 存储:至少 500 GB 可用空间
- 操作系统:Linux,推荐使用 Ubuntu 18.04+ 或 CentOS 7+
使用 Nextflow 运行工作流
Nextflow 是推荐的用于批量处理、报告生成和工作流管理的方法。
安装 Nextflow
bash
conda install -n seeksoulmethyl -c bioconda nextflow运行主工作流
bash
nextflow run -bg SeekSoulMethyl/nf/main.nf \
--outdir /path/to/results/ \
--samplesheet samplelist.csv \
-w /path/to/results/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/human-reference-GRCh38/ \
--split_fastq 1 \
--filter_ch 2 \
--chemistry DD-MET3 > methy.log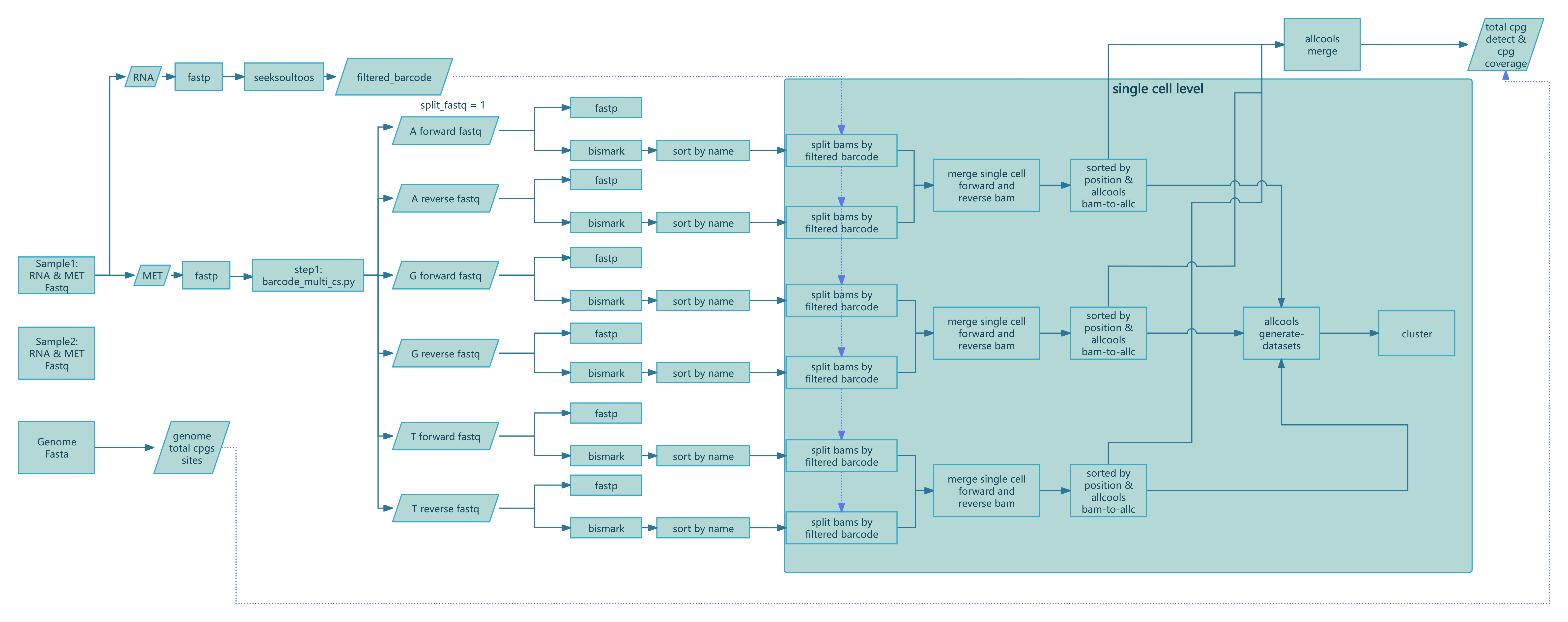
图 1:SeekSoul™ Methyl Tools Nextflow 流程图
支持的 Nextflow 工作流
v2.1.2 代码库通过 --workflow 支持多个工作流:
rna_met:转录组加甲基化综合分析methy_only:仅甲基化工作流force_cell:基于之前的输出重新计算或更新甲基化结果
仅甲基化工作流
bash
nextflow run SeekSoulMethyl/nf/main.nf \
--workflow methy_only \
--outdir /path/to/results \
--samplesheet samplelist.csv \
-w /path/to/work \
-c SeekSoulMethyl/nf/nextflow.config \
-profile aliyun_k8s \
--database_dir /path/to/reference \
--split_fastq 4 \
--filter_ch 2 \
--chemistry DD-MET3有关 nextflow.config 的说明
文件 nf/nextflow.config 必须根据您的基础架构进行调整。请关注以下项目:
process.executor:选择local、slurm、pbs、k8s、awsbatch或其他支持的执行器process.cpus、process.memory、process.time:默认资源限制workDir:具有足够可写存储空间的工作目录conda.enabled、docker.enabled或singularity.enabled:选择您的平台支持的环境策略
典型的 profile 包括本地执行、Slurm 集群和 Kubernetes 环境。
关键运行时参数
--database_dir:参考基因组数据库目录--chemistry:选择DD-MET3或DD-MET5--split_fastq:根据前n个 barcode 碱基拆分甲基化 FASTQ 文件以提高并行度--filter_ch:移除包含超过n个 CH 甲基化位点的 read 对;设置为0可禁用-resume:使用相同的工作目录重新启动失败或中断的 Nextflow 运行
常见问题
- Samplesheet 解析错误:确认第一列恰好是
sample_id,并使用绝对文件路径。 - 缺少
${sample}.mcds:检查是否生成了每个细胞的*_allc.gz文件,并确认染色体大小资源是否有效。 - Bismark 比对停止或失败:确认在
--database_dir/fasta/下存在 Bismark 参考基因组,并且对执行环境可见。 - 重复运行:保持相同的
-w工作目录并添加-resume。