Algorithm
Data Structure
SeekOne DD Single Cell Multiome Methylation + RNA libraries are provided in two chemistries.
DD-MET3 Methylation Library Structure
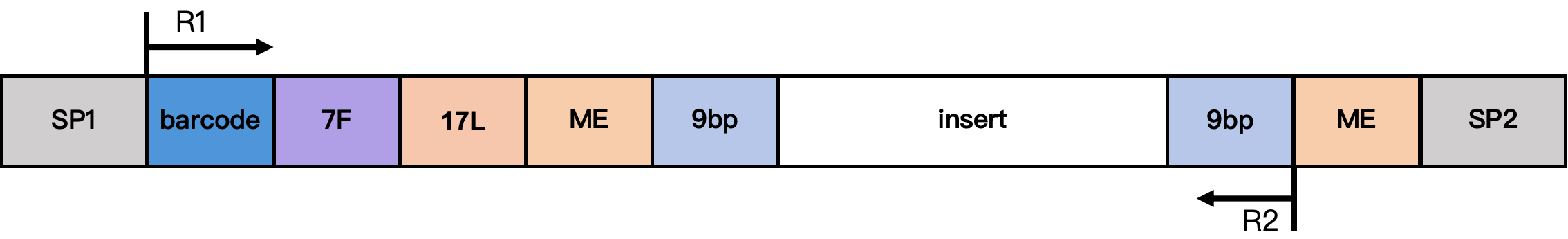
Structure notes:
- SP1/SP2: adapter sequences
- barcode: 17 bp cell barcode
- 7F: 7 bp linker sequence
- 17L: 17 bp fixed sequence
CgtCCgtCgttgCtCgt - ME: 19 bp fixed sequence
AGATGTGTATAAGAGACAG - 9 bp: extension sequence from the Tn5 insertion fragment
DD-MET5 Methylation Library Structure
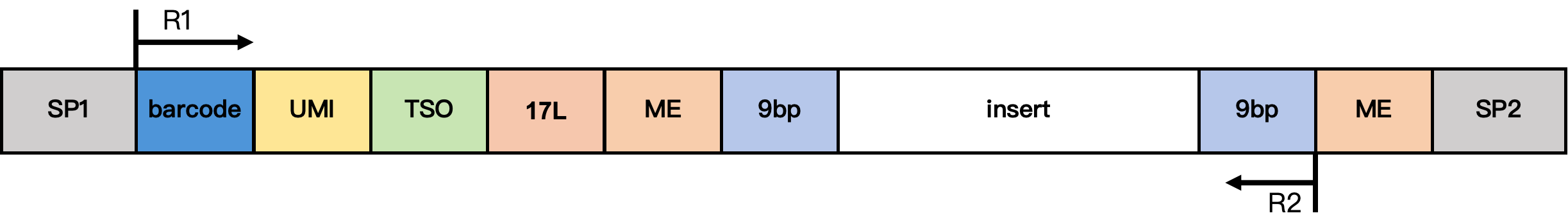
Structure notes:
- SP1/SP2: adapter sequences
- barcode: 17 bp cell barcode
- UMI: 12 bp UMI sequence
- TSO: 13 bp TSO sequence
TTTCTTATATGGG - 17L: 17 bp fixed sequence
CgtCCgtCgttgCtCgt - ME: 19 bp fixed sequence
AGATGTGTATAAGAGACAG - 9 bp: extension sequence from the Tn5 insertion fragment
During enzymatic conversion, unmethylated cytosines are converted to thymines. Adapter cytosines are protected, while specific fixed-sequence cytosines are intentionally left unprotected and are later used to estimate the C-to-T conversion rate.
Transcriptome Workflow
Transcriptome processing is performed through SeekSoulTools. Cells retained for methylation downstream analysis are determined using transcriptome-derived cell barcodes.
Methylation Workflow
Step 1: Preprocessing and Barcode Parsing
Barcode extraction and correction
The pipeline locates the barcode region using the expected library structure. If the extracted barcode appears in the whitelist, it is kept directly. Otherwise, the pipeline attempts one-mismatch barcode correction:
- If exactly one whitelist candidate is found, the barcode is corrected to that candidate.
- If several candidates match, the barcode supported by the highest read count is selected.
- If no valid correction is found, the read is discarded as an invalid-barcode read.
UMI extraction
UMIs are extracted from predefined positions and are not corrected during parsing.
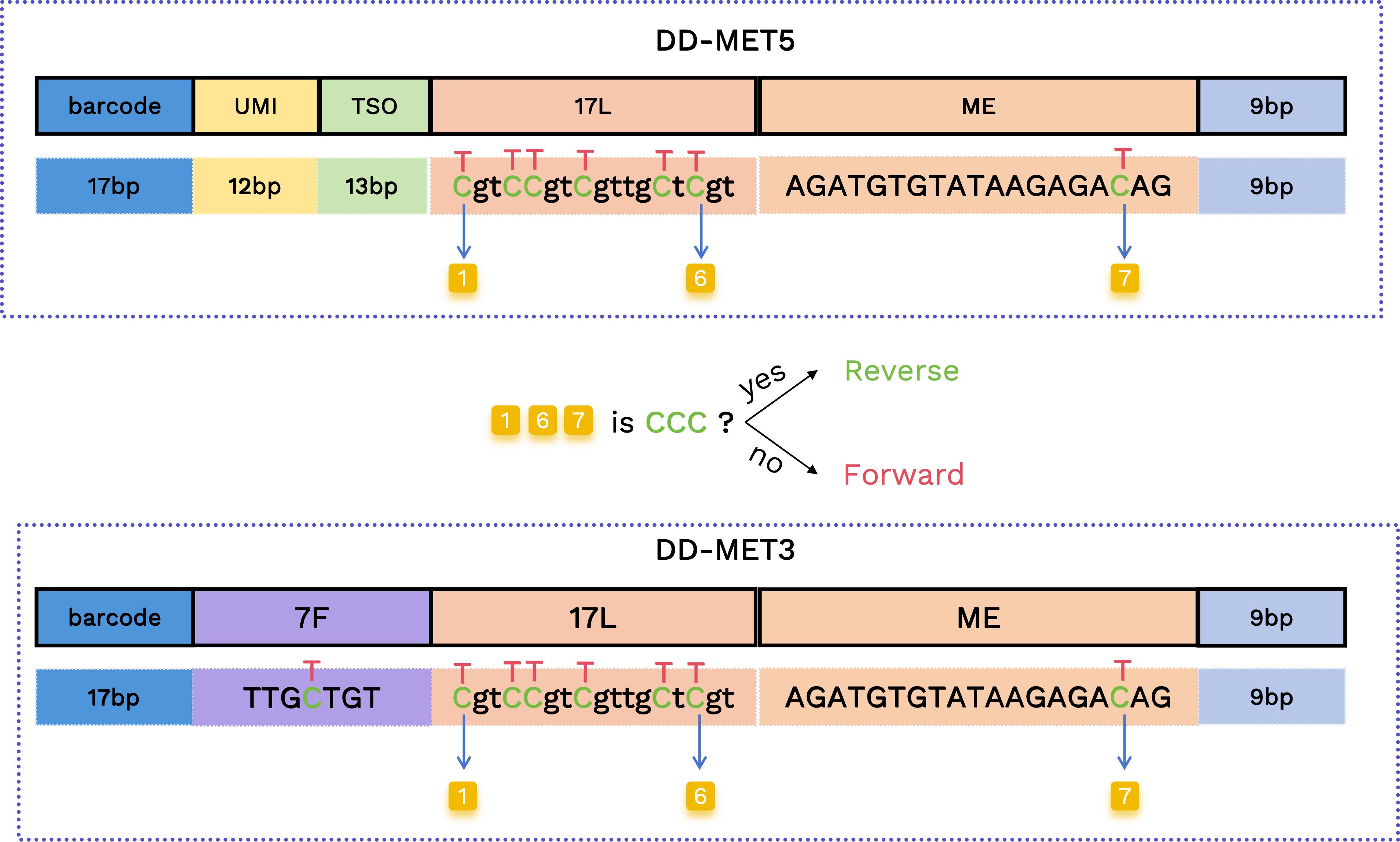
Forward and reverse read determination
The first and last two original cytosine positions in the 17L and ME segments are used to distinguish forward and reverse reads. If all three positions remain cytosines, the read is classified as reverse; otherwise it is classified as forward.
- Forward read pattern:
TgtTTgtTgttgTtTgtAGATGTGTATAAGAGAT - Reverse read pattern:
CgtCCgtCgttgCtCgtAGATGTGTATAAGAGAC
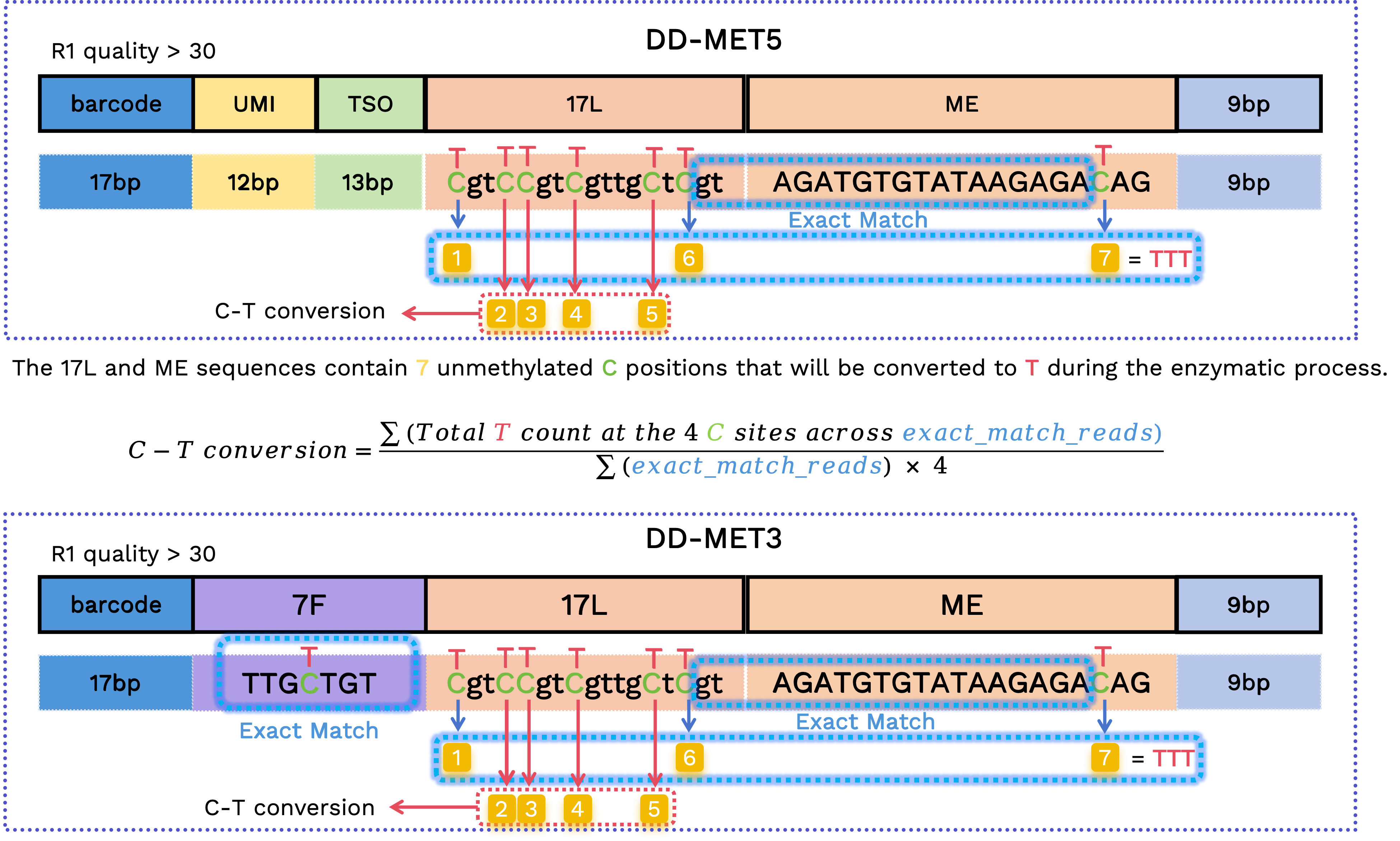
C-to-T conversion rate
To estimate conversion efficiency, the workflow inspects fixed-sequence cytosine sites in the 17L and ME segments, but only for reads with verified structures. This calculation is restricted to a high-confidence subset and does not remove reads from the final FASTQ outputs.
Read cleanup and filtering
The pipeline then removes artificial library segments, trims overlapping adapters, trims the 9 bp Tn5 gap regions, optionally filters reads with excessive non-CpG methylation, and removes reads that become too short after trimming.
Step 2: Bismark Alignment and BAM Sorting
A customized Bismark build is used for methylation alignment. The workflow adds corrected cell barcodes and raw UMIs to BAM tags, aligns forward and reverse read groups with different options, and sorts BAM files by read name for downstream processing.
Step 3: ALLCools Analysis
The workflow splits name-sorted BAM files by cell barcode, converts each per-cell BAM into ALLC format, performs UR-tag-based UMI handling with the customized ALLCools implementation, and builds multi-scale methylation datasets such as chrom10k, chrom20k, chrom50k, chrom100k, chrom500k, chrom1M, and geneslop2k.
Step 4: Dimensionality Reduction and Visualization
By default, dimensionality reduction is performed on chrom20k bins with LSI, followed by UMAP visualization.
Workflow Summary
At the implementation level, the Nextflow pipeline is organized into four major process groups:
step1: QC, transcriptome analysis, methylation barcode parsing, and FASTQ shardingstep2: Bismark alignment and BAM name sortingstep3: per-cell BAM splitting, ALLC generation, ALLC merge, and MCDS generationstep4: summary statistics, clustering, visualization, and integrated HTML reporting
Nextflow Step-by-Step Details
This section captures the detailed process-oriented description from the README and maps it back into the documentation structure used here.
Step 1: Preprocessing and Barcode Parsing
COMPUTE_CPG_SITES: counts genome-wide CpG sites fromgenome.faand chromosome size informationFASTP_EXPRESSION_MULTI: performs transcriptome FASTQ trimming and QCFASTP_METHYLATION_MULTI: performs methylation FASTQ QC before barcode parsingSEEKSOULTOOLS_RNA: runs transcriptome alignment, counting, filtering, clustering, and differential expressionMETHYLATION_BARCODE_EXTRACTION: parses methylation barcodes and UMIs according to chemistry type, performs barcode correction, and can shard FASTQs with--split_fastqPARSE_FASTQ_FILES: pairs forward and reverse FASTQ fragments for downstream mappingFASTP_METHYLATION_BARCODE_EXTRACT: runs post-barcode-extraction QC on the parsed methylation FASTQs
Step 2: Bismark Alignment and BAM Sorting
BISMARK_ALIGNMENT_FORWARD: aligns forward methylation reads with barcode and UMI tagsBISMARK_ALIGNMENT_REVERSE: aligns reverse methylation reads with PBAT mode enabledSORT_BAM_BY_NAME: sorts BAM files by read name so downstream per-cell splitting can be performed correctly
Step 3: Per-cell Split, ALLC Generation, Merge, and Dataset Building
SPLIT_BAM_FILES: splits name-sorted BAMs into per-cell BAM files using transcriptome-derived barcodesMERGE_BISMARK_BAM: merges forward and reverse per-cell BAMs and combines barcode count summariesALLCOOLS_BAM_TO_ALLC: converts per-cell BAMs to ALLC format using the customized ALLCools implementationMERGE_FILTERED_BARCODE_READS_COUNTS: merges barcode-level metrics and writes consolidated cell summariesALLCOOLS_GENERATE_DATASETS: builds multi-scale methylation datasets such as chrom10k, chrom20k, chrom50k, chrom100k, chrom500k, chrom1M, and geneslop2kALLCOOLS_SUBMERGEandALLCOOLS_MERGE: merge sharded ALLC outputs when FASTQ splitting is enabledALLCOOLS_EXTRACT: extracts merged CG-context ALLC outputs for downstream analysis
For the methy_only workflow, read-count-based barcode estimation and filtering are additionally handled through helper utilities in utils.nf.
Step 4: Summary, Dimensionality Reduction, and Joint Report
METHYLATION_SUMMARY: aggregates Bismark reports, cell metrics, and CpG statistics into JSON and CSV summariesMETHYLATION_LSI_PCA_CLUSTERING: performs dimensionality reduction and clustering, using LSI on chrom20k bins by defaultMULTI_REPORT: combines transcriptome and methylation outputs into the integrated HTML report
References
[1] Lu X, Yuan Y, et al. Improved tagmentation-based whole-genome bisulfite sequencing for input DNA from less than 100 mammalian cells. Epigenomics. 2015;7(1):47-56. doi:10.2217/epi.14.76.