算法原理
数据结构
SeekOne DD Single Cell Multiome Methylation + RNA 文库提供两种化学方法。
DD-MET3 甲基化文库结构
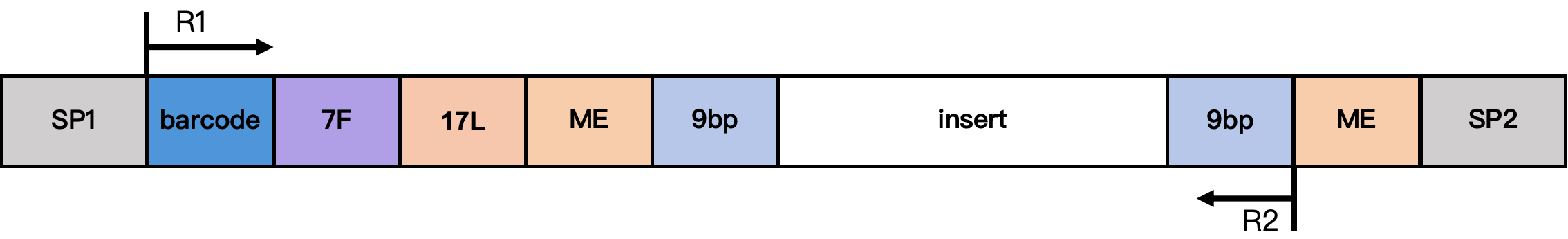
结构说明:
- SP1/SP2:接头序列
- barcode:17 bp 细胞 barcode
- 7F:7 bp 连接序列
- 17L:17 bp 固定序列
CgtCCgtCgttgCtCgt - ME:19 bp 固定序列
AGATGTGTATAAGAGACAG - 9 bp:Tn5 插入片段的延伸序列
DD-MET5 甲基化文库结构
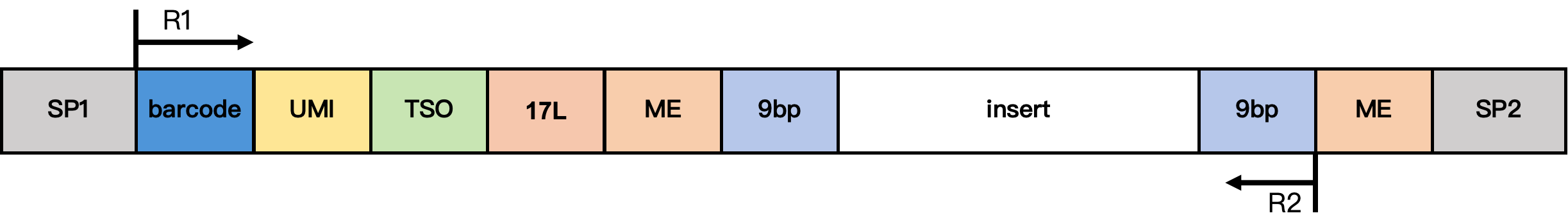
结构说明:
- SP1/SP2:接头序列
- barcode:17 bp 细胞 barcode
- UMI:12 bp UMI 序列
- TSO:13 bp TSO 序列
TTTCTTATATGGG - 17L:17 bp 固定序列
CgtCCgtCgttgCtCgt - ME:19 bp 固定序列
AGATGTGTATAAGAGACAG - 9 bp:Tn5 插入片段的延伸序列
在酶转化过程中,未甲基化的胞嘧啶被转化为胸腺嘧啶。接头胞嘧啶受到保护,而特定的固定序列胞嘧啶则故意不加保护,稍后用于估计 C 到 T 的转化率。
转录组工作流
转录组处理通过 SeekSoulTools 执行。根据转录组派生的细胞 barcode 确定保留用于甲基化下游分析的细胞。
甲基化工作流
第 1 步:预处理和 Barcode 解析
Barcode 提取和校正
流程根据预期的文库结构定位 barcode 区域。如果提取的 barcode 出现在白名单中,则直接保留。否则,流程将尝试进行一个错配的 barcode 校正:
- 如果恰好找到一个白名单候选者,则将 barcode 校正为该候选者。
- 如果匹配到多个候选者,则选择 read 计数最高的 barcode。
- 如果未找到有效的校正,则将该 read 作为无效 barcode read 丢弃。
UMI 提取
从预定义位置提取 UMI,在解析期间不对其进行校正。
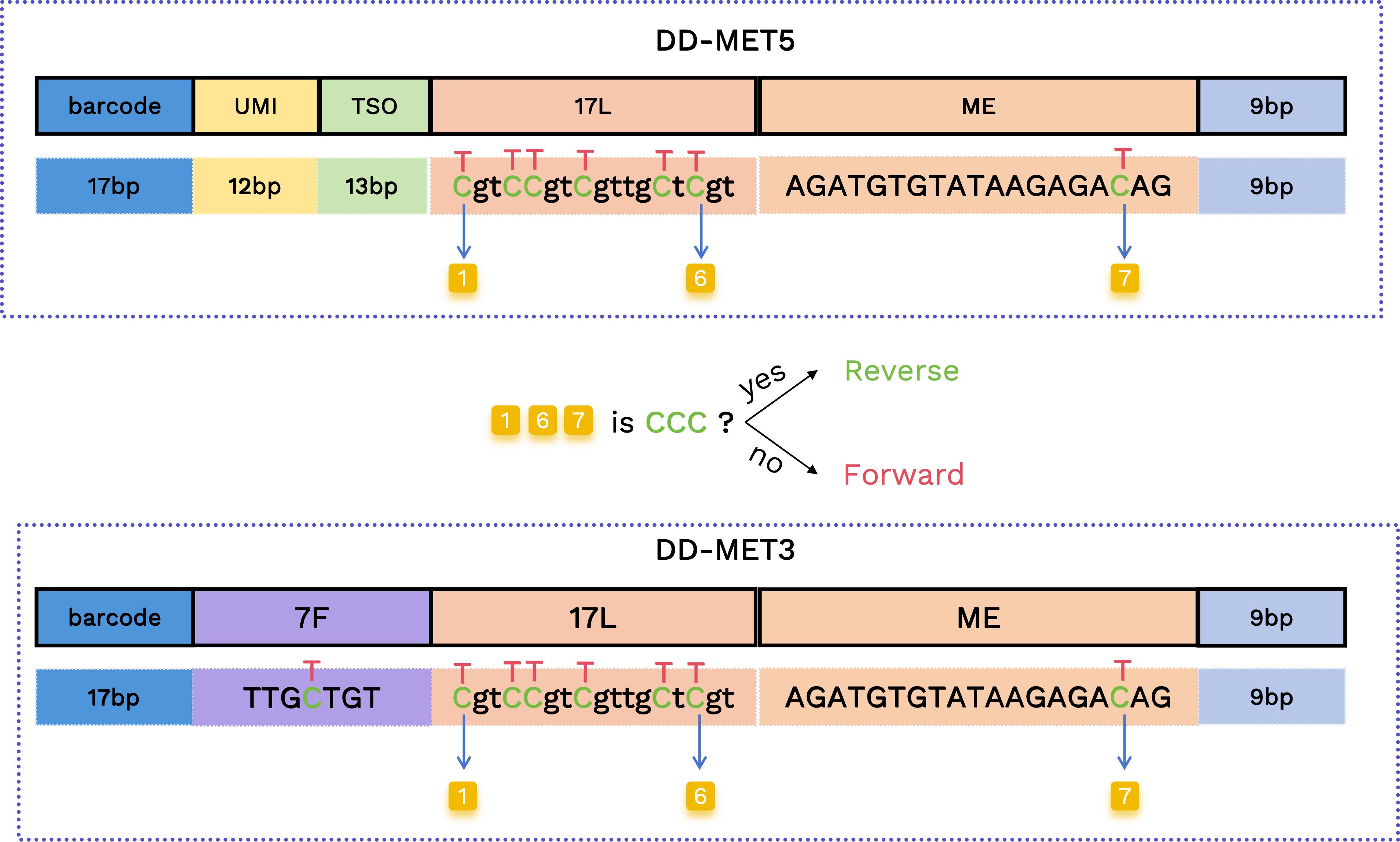
正向和反向 read 确定
使用 17L 和 ME 片段中的前两个和最后一个原始胞嘧啶位置来区分正向和反向 read。如果这三个位置都保留为胞嘧啶,则将该 read 分类为反向;否则分类为正向。
- 正向 read 模式:
TgtTTgtTgttgTtTgtAGATGTGTATAAGAGAT - 反向 read 模式:
CgtCCgtCgttgCtCgtAGATGTGTATAAGAGAC
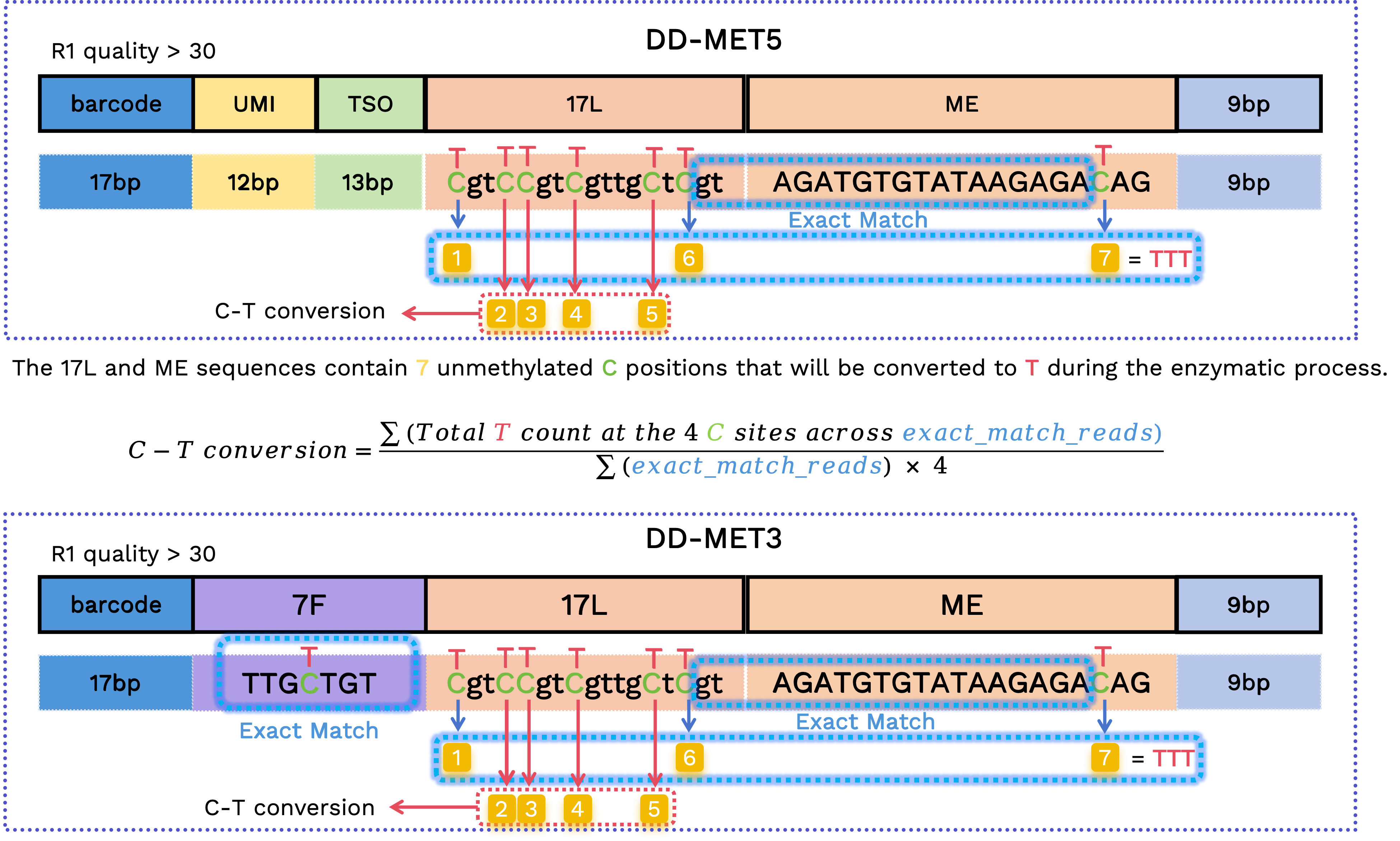
C 到 T 转化率
为了估计转化效率,工作流检查 17L 和 ME 片段中的固定序列胞嘧啶位点,但仅针对结构经验证的 read。此计算仅限于高置信度子集,并且不会从最终的 FASTQ 输出中删除 read。
Read 清理和过滤
随后,流程会移除人工文库片段,修剪重叠接头,修剪 9 bp Tn5 缺口区域,可选择过滤具有过多非 CpG 甲基化的 read,并移除修剪后变得太短的 read。
第 2 步:Bismark 比对和 BAM 排序
甲基化比对使用定制的 Bismark 构建。工作流将校正后的细胞 barcode 和原始 UMI 添加到 BAM 标签中,使用不同的选项比对正向和反向 read 组,并按 read 名称对 BAM 文件进行排序,以供下游处理。
第 3 步:ALLCools 分析
工作流按细胞 barcode 拆分按名称排序的 BAM 文件,将每个细胞的 BAM 转换为 ALLC 格式,使用定制的 ALLCools 实现执行基于 UR 标签的 UMI 处理,并构建多尺度甲基化数据集,例如 chrom10k、chrom20k、chrom50k、chrom100k、chrom500k、chrom1M 和 geneslop2k。
第 4 步:降维和可视化
默认情况下,对 chrom20k bin 执行 LSI 降维,然后进行 UMAP 可视化。
工作流摘要
在实现层面,Nextflow 流程分为四个主要的进程组:
step1:QC、转录组分析、甲基化 barcode 解析和 FASTQ 分片step2:Bismark 比对和 BAM 按名称排序step3:单细胞拆分、ALLC 生成、ALLC 合并和 MCDS 生成step4:摘要统计、聚类、可视化和综合 HTML 报告
Nextflow 详细步骤说明
本节捕获了自述文件中面向过程的详细描述,并将其映射回此处使用的文档结构中。
第 1 步:预处理和 Barcode 解析
COMPUTE_CPG_SITES:从genome.fa和染色体大小信息计算全基因组 CpG 位点FASTP_EXPRESSION_MULTI:执行转录组 FASTQ 修剪和 QCFASTP_METHYLATION_MULTI:在 barcode 解析前执行甲基化 FASTQ QCSEEKSOULTOOLS_RNA:运行转录组比对、计数、过滤、聚类和差异表达METHYLATION_BARCODE_EXTRACTION:根据化学类型解析甲基化 barcode 和 UMI,执行 barcode 校正,并可使用--split_fastq对 FASTQ 进行分片PARSE_FASTQ_FILES:将正向和反向 FASTQ 片段配对以进行下游比对FASTP_METHYLATION_BARCODE_EXTRACT:对解析后的甲基化 FASTQ 执行 barcode 提取后的 QC
第 2 步:Bismark 比对和 BAM 排序
BISMARK_ALIGNMENT_FORWARD:比对带有 barcode 和 UMI 标签的正向甲基化 readBISMARK_ALIGNMENT_REVERSE:在启用 PBAT 模式的情况下比对反向甲基化 readSORT_BAM_BY_NAME:按 read 名称对 BAM 文件进行排序,以便正确执行下游的单细胞拆分
第 3 步:单细胞拆分、ALLC 生成、合并和数据集构建
SPLIT_BAM_FILES:使用转录组派生的 barcode 将按名称排序的 BAM 拆分为单细胞 BAM 文件MERGE_BISMARK_BAM:合并正向和反向的单细胞 BAM,并组合 barcode 计数摘要ALLCOOLS_BAM_TO_ALLC:使用定制的 ALLCools 实现将单细胞 BAM 转换为 ALLC 格式MERGE_FILTERED_BARCODE_READS_COUNTS:合并 barcode 级别的指标并编写综合的细胞摘要ALLCOOLS_GENERATE_DATASETS:构建多尺度甲基化数据集,例如 chrom10k、chrom20k、chrom50k、chrom100k、chrom500k、chrom1M 和 geneslop2kALLCOOLS_SUBMERGE和ALLCOOLS_MERGE:启用 FASTQ 拆分时合并分片的 ALLC 输出ALLCOOLS_EXTRACT:提取合并的 CG 上下文 ALLC 输出用于下游分析
对于 methy_only 工作流,基于 read 计数的 barcode 估计和过滤会额外通过 utils.nf 中的辅助工具进行处理。
第 4 步:摘要、降维和联合报告
METHYLATION_SUMMARY:将 Bismark 报告、细胞指标和 CpG 统计数据汇总为 JSON 和 CSV 摘要METHYLATION_LSI_PCA_CLUSTERING:执行降维和聚类,默认对 chrom20k bin 使用 LSIMULTI_REPORT:将转录组和甲基化输出组合成综合 HTML 报告
参考文献
[1] Lu X, Yuan Y, et al. Improved tagmentation-based whole-genome bisulfite sequencing for input DNA from less than 100 mammalian cells. Epigenomics. 2015;7(1):47-56. doi:10.2217/epi.14.76.