甲基化 + RNA 双组学分析指南
单细胞转录组与单细胞甲基化可在同一细胞内同步获取基因表达与表观遗传信息,帮助解析“甲基化变化—调控因子—基因表达—细胞表型”之间的关联关系。
基于甲基化 + RNA 双组学数据,可以从细胞类型注释、亚群识别、发育或疾病状态变化、肿瘤异质性与克隆结构等多个层面进行系统分析。同时,通过差异甲基化区域(DMR)、差异甲基化基因(DMG)、差异表达基因(DEG)、功能富集和 motif 分析等结果,可进一步构建“表观调控—基因表达—生物学功能”的证据链,为机制解释、候选靶点筛选和后续实验验证提供参考。
为帮助用户快速理解单细胞甲基化双组学的分析思路与关键产出,寻因生物提供系统化的下游分析流程与教程,覆盖双细胞判定、数据质控与整合、差异表达基因(DEG)分析、差异甲基化区域(DMR)分析、功能富集与 motif 分析、甲基化谱系与克隆结构分析、肿瘤细胞判定等关键环节。
每个教程均明确输入文件规范、适用场景、核心分析步骤和主要输出结果,并配有标准化代码与示例结果,便于用户根据自身项目需求选择合适的分析模块,完成数据解读与结果展示。
SeekSoul Methyl Tools
SeekSoul Methyl Tools 是寻因生物面向单细胞甲基化双组学的标准化分析流程,支持从原始测序数据 (FASTQ) 到细胞层面的表达与甲基化检出的全链路处理。一站式覆盖质控、reads 比对、细胞表达矩阵构建、甲基化位点检测、结果可视化与报告生成等关键环节;同时提供完整的运行环境要求、安装指南、示例数据与参数模板,便于快速上手与批量项目部署,构建从“原始数据”到“可分析矩阵”的稳定、可复用生产线。
🔗 SeekSoul Methyl Tools 网址:SeekSoul Methyl Tools
双细胞鉴定
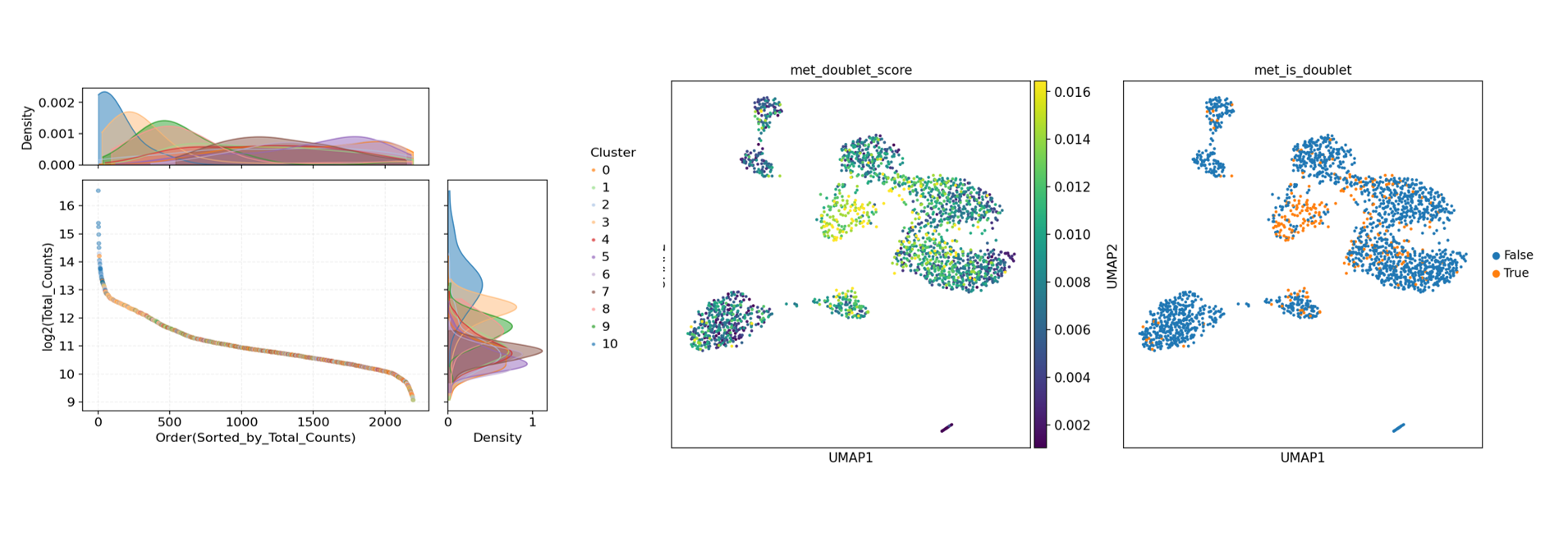
单细胞建库可能因上样过浓、细胞黏连/受损、液滴同封两细胞、条形码/库构建过程的索引碰撞等产生”双细胞 (doublet)”,其表达或甲基化特征呈混合。若不剔除,易制造伪中间态与伪聚类,干扰细胞类型注释,降低结果的可解释性与复现性。因此,应在下游分析前对双细胞进行识别与过滤。
寻因生物提供的双细胞识别教程:甲基化+RNA双组学-双细胞判断.ipynb
该教程综合三类双细胞判定方法:
- 基于转录组的表达异常。
- 基于甲基化 reads 数的异常。
- 基于甲基化率的 MethylScrublet 判定。
从不同维度提高双细胞检测的稳健性。
输入文件
- 转录组表达矩阵:RNA 水平基因表达矩阵(
filtered_feature_bc_matrix文件夹)。 - 单细胞甲基化数据:MCDS 格式的甲基化数据集(
.mcds文件)。 - Barcode 映射文件(可选):如果是双标签文库,即使用 DD-MET3 建库,需将 RNA 和甲基化数据的细胞 Barcode 进行对应。
- 下载地址:DD-M_bUCB3_whitelist.csv。
输出文件
- 双细胞判定结果:
sample_doublet.txt。 - 可视化图件:细胞 UMI 可视化分布图、甲基化 reads 分布图、双细胞鉴定分布图等。
基础分析
在单细胞分析中,需要将不同样品、分组与批次的数据投射到统一坐标系进行对比。样品整合不仅能提升细胞类型注释的一致性、增强稀有细胞与弱信号的检出能力,还可以支持跨样品与跨条件(如不同患者、不同处理)比较,构建可扩展的参考图谱,并稳健识别跨数据集中复现的细胞群,从而推动后续数据的深入挖掘。
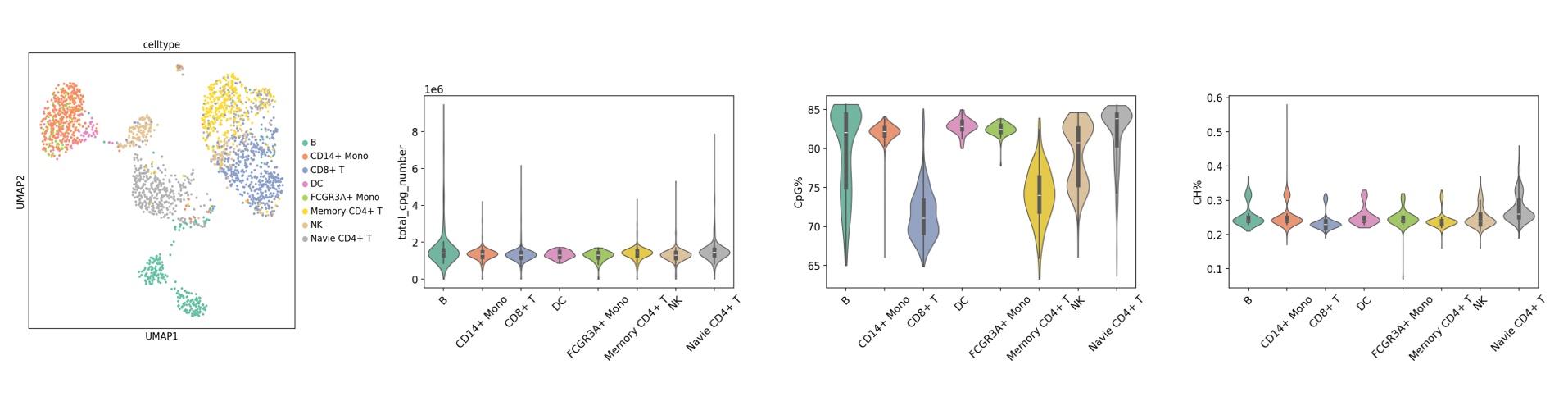
基础分析教程:甲基化+RNA双组学-基础分析.ipynb
该教程是寻因生物基于 ALLCools、Scanpy、Harmony 开发的甲基化双组学整合流程模板,涵盖数据质控、批次校正、降维聚类与可视化。用户按模板配置输入与参数,即可快速完成整合分析,获得标准化、可复用的结果与图件,提高分析效率与结论的可解释性与复现性。
输入文件
- 转录组表达矩阵:RNA 水平基因表达矩阵(
filtered_feature_bc_matrix文件夹)。 - 单细胞甲基化数据:MCDS 格式的甲基化数据集(
.mcds文件)。 - Barcode 映射文件(可选):如果是双标签文库,即使用 DD-MET3 建库,需将 RNA 和甲基化数据的细胞 Barcode 进行对应。
- 下载地址:DD-M_bUCB3_whitelist.csv。
输出文件
adata_rna.h5ad:表达数据整合结果。adata_met.h5ad:甲基化数据整合结果。- 可视化图片:质控图、聚类 UMAP 图、基因表达 UMAP 等。
多模态整合分析
在完成单细胞转录组 (RNA) 与单细胞甲基化 (DNA Methylation) 的基础分析后,如何将这两种不同维度的数据在同一细胞层面进行深度整合,是揭示表观遗传调控机制的关键。通过多模态整合,可以突破单一组学的局限,更精准地定义细胞状态,并解析从 DNA 甲基化修饰到基因表达水平的调控逻辑。
寻因生物提供以下两种主流的多模态整合方案:
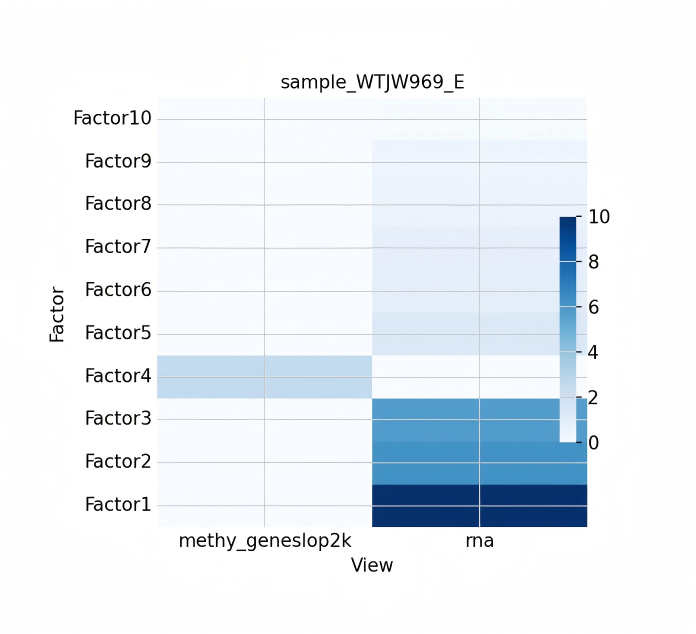
- MOFA+ 多模态整合分析。 教程文档:甲基化 + RNA 双组学_MOFA+ 多模态整合分析.ipynb。 使用 MOFA+ (Multi-Omics Factor Analysis) 框架以无监督学习方式整合单细胞 RNA 与甲基化数据。通过潜在因子 (Latent Factors) 分析,该方法能够有效量化不同组学对细胞异质性的贡献权重,识别跨组学的共有变异与组学特异性变异,非常适合探索复杂的组学间相互作用并发现新的细胞亚群特征。
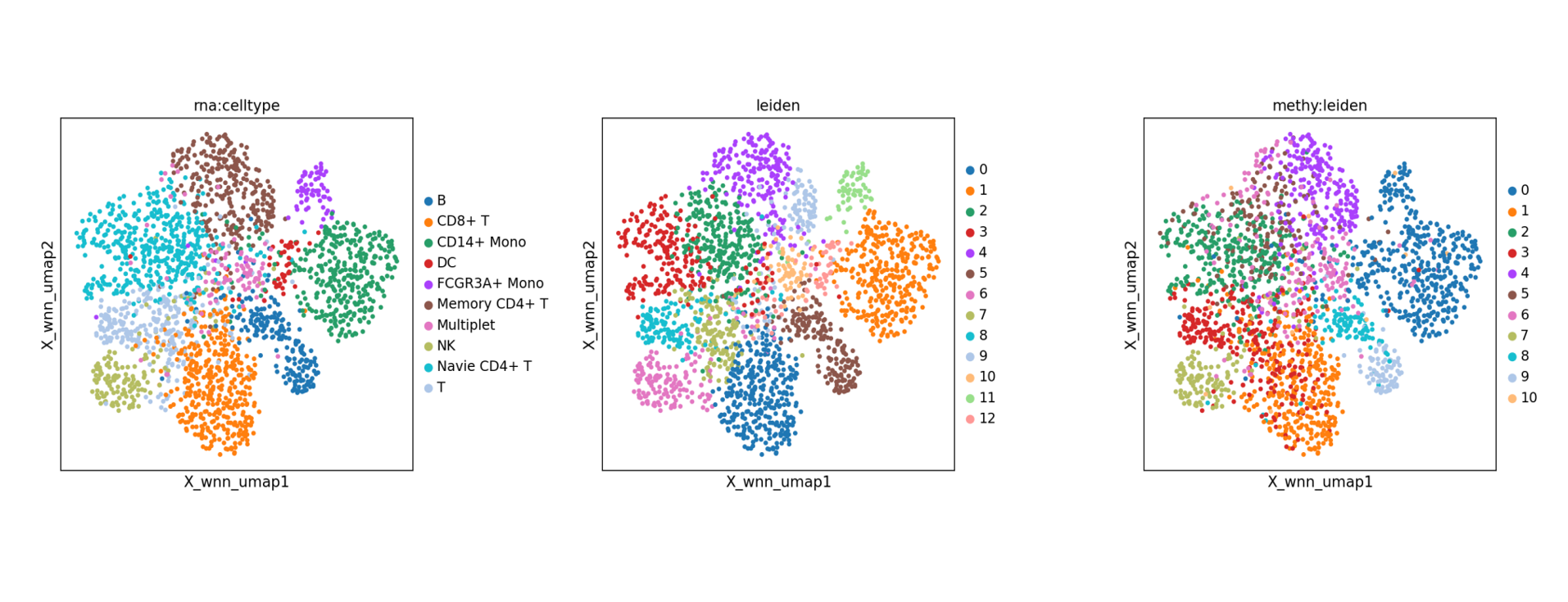
- WNN 多模态整合分析。 教程文档:甲基化 + RNA 双组学_WNN 多模态整合分析.ipynb。 基于 Seurat 框架下的 WNN (Weighted Nearest Neighbor) 算法,根据每个细胞中各模态的信息含量自动分配权重并构建加权最近邻图。该方法同时结合了 RNA 的高灵敏度与甲基化的表观特征,实现了更稳健的聚类与可视化分析。相比单一组学,WNN 整合通常具有更高的生物学分辨率,能够识别出在单组学中被掩盖的细微细胞状态差异。
差异与功能富集
单细胞注释完成后,需系统比较不同细胞群/亚群的分子差异,产出可解释的 DEG/DMG/DMR 清单,并将“差异基因/区域”映射到生物过程与通路,识别关键调控因子(TF)与核心机制,从海量差异中提炼高价值功能模块,定位用于诊断/分型/预后的候选通路与基因,支撑靶点与标志物筛选及疗效/免疫评估。同时,将甲基化(DMR/DMG)与表达(DEG)置于统一的功能框架进行关联,构建“表观—表达—表型”的证据链,提升结论的可信度与可转化性。
差异分析教程:甲基化+RNA双组学-差异分析和功能富集分析.ipynb
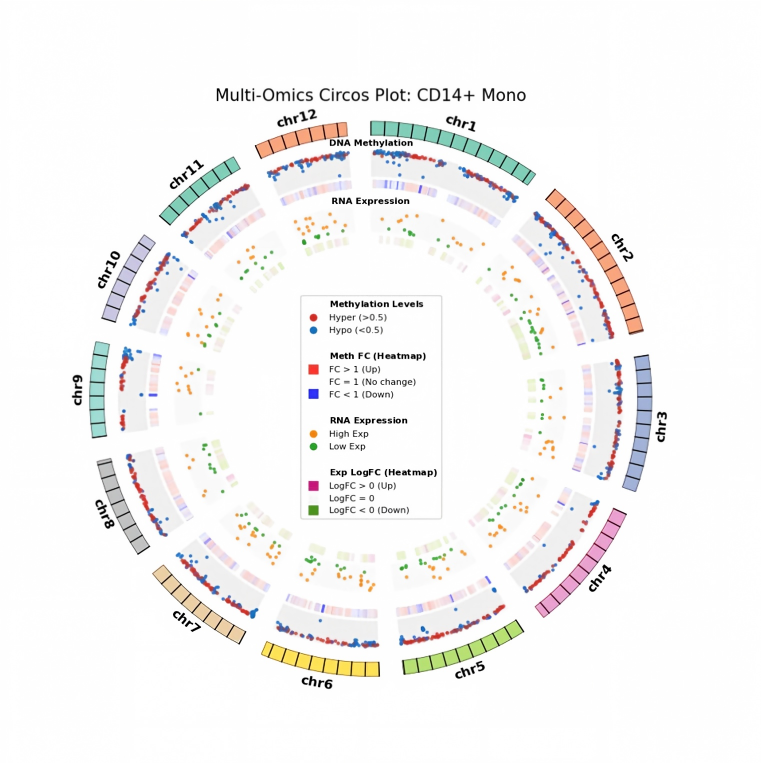
基于甲基化双组学数据的细胞群间差异功能富集分析流程。在完成细胞注释的前提下,以基因组 20 kb 窗口进行 DMB 分析、以基因区 ±2 kb 进行 DMG 分析,并识别 DEG;随后通过 Venn 与 Circos 展示多组学可视化关联分析,解析甲基化水平与基因表达的联动关系,形成机制线索与优先验证名单。
输入文件
adata_rna.h5ad:RNA 水平整合注释文件,包含细胞注释信息和基因 ID,即 h5ad 包含obs['celltype']、var['gene_ids']。adata_met.h5ad:甲基化水平整合注释文件,包含注释的甲基化数据和样品信息,即 h5ad 包含obs['celltype']、obs['Sample']。- MCDS 格式数据:包含 20 kb 窗口和基因区域 ±2 kb 的甲基化信息,即
chrom20k、geneslop2k;如果想用其他区域数据,可自行设置。
输出文件
- DMB 分析结果:不同细胞群差异甲基化块分析结果。
DEG.csv:不同细胞群差异基因分析结果。hdf.hdf文件:所有细胞群的 DMG、DMB 分析结果。- 可视化图:DEG 与 DMG 可视化 Venn 图、Circos 图、GO/KEGG 可视化条形图、气泡图等。
组间差异分析
单细胞注释完成后,除比较不同组别(如疾病/对照、处理/未处理)在各细胞群的组成与占比差异外,更需系统评估组间在细胞群内的基因表达与甲基化变化,将检测到的差异与潜在生物学机制关联,识别驱动细胞状态转换的表观遗传调控因子与信号通路。通过将甲基化与转录组数据联动并锚定到具体细胞群,定位关键细胞类型、通路与调控元件,为理解表观遗传改变如何影响基因表达并最终导致发育或疾病等表型差异提供直接的机制性假说。
组间差异分析教程:甲基化+RNA双组学-组间细胞类型差异富集分析.ipynb
基于甲基化双组学的组间差异分析教程:在细胞注释基础上,按细胞类型开展差异表达基因 (DEG) 与差异甲基化基因 (DMG) 分析,并对差异基因进行 GO/KEGG 功能富集;随后进行多组学关联整合(如 DEG 与 DMG 的交叉分析),解析甲基化水平与基因表达的关系,并以 Venn 与 Circos 图直观展示双组学关联;同时挖掘关键转录因子,阐释表观遗传修饰对基因表达的调控作用。
输入文件
adata_rna.h5ad:RNA 水平整合注释文件,包含细胞注释信息和基因 ID,即 h5ad 包含obs['celltype']、var['gene_ids']。adata_met.h5ad:甲基化水平整合注释文件,包含注释的甲基化数据和样品信息,即 h5ad 包含obs['celltype']、obs['Sample']。- MCDS 格式数据:包含基因区域 ±2 kb 的甲基化信息,即
geneslop2k;如果想用其他区域数据,可自行设置。
输出文件
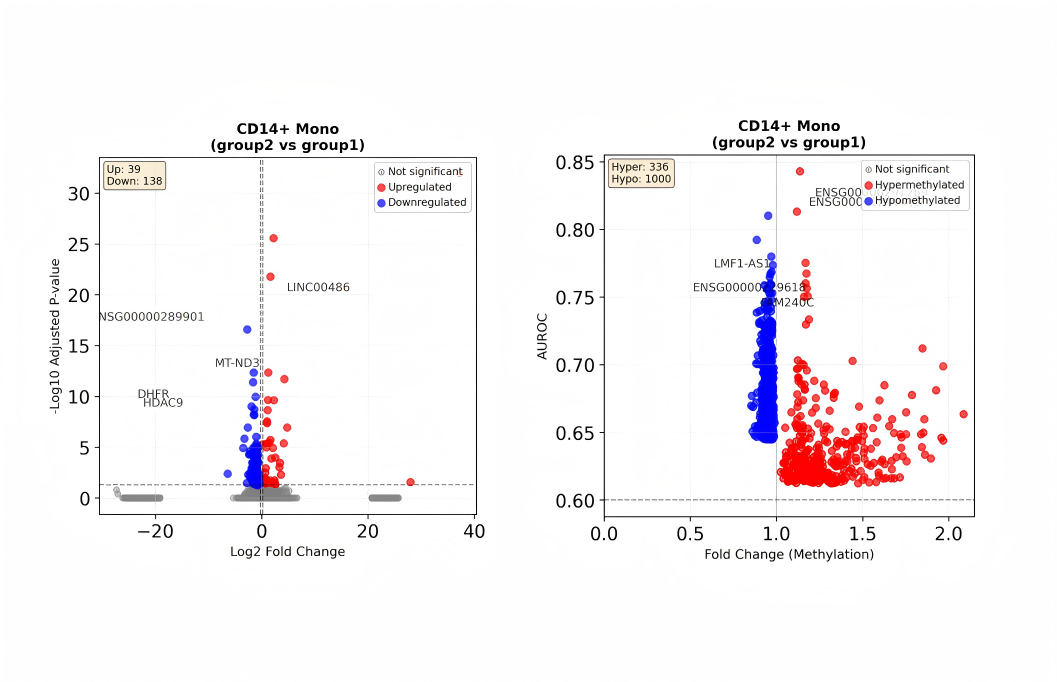
DEG_group_comparison.csv:细胞群不同组间差异分析结果。DMG_group_comparison.csv:细胞群不同组间 DMG 差异分析结果。- 可视化图:DEG 火山图、DMG 差异火山图、DEG 与 DMG 关联可视化 Venn 图、Circos 图、GO/KEGG 可视化条形图、气泡图等。
MethSCAn 分析
MethSCAn 是用于单细胞甲基化数据的分析工具。其核心流程包括:将单细胞甲基化原始读段数转化为按染色体坐标的稀疏矩阵 (CSR);沿基因组对位点信号进行平滑以降低低覆盖与位置随机性带来的噪声;以细胞间甲基化变异为准则扫描全基因组,检测变异甲基化区域 (VMR);并在区域层面进行两组比较,输出差异甲基化区域 (DMR)。
MethSCAn 教程:由寻因生物提供,基于单细胞甲基化数据,使用 MethSCAn 完成 VMR 检测与 DMR 分析。
- 文档说明:甲基化+RNA双组学-MethSCAn差异甲基化分析.ipynb。
- 格式转换:在运行前,需将 SeekSoulMethyl 流程产生的 allc 文件转换为 bismark coverage 格式。
- 转换脚本:allc_to_bismarkCov.py。
DMRs_rGREAT 分析
在完成单细胞甲基化数据的差异甲基化区域 (DMR) 分析后,需将 DMR 关联到邻近基因并进行功能富集(甲基化区域富集分析),以明确其可能影响的生物学过程与信号通路。从大量区域中归纳关键 GO/KEGG 条目、候选调控链路与潜在驱动因子,并将 DMR 与 DEG/转录因子 (TF) 富集结果统一到同一功能框架,构建“表观—表达—表型”的连贯证据链。
rGREAT 分析教程:甲基化+RNA双组学-差异甲基化区域功能富集分析.ipynb
寻因生物采用 rGREAT 对 DMR 进行注释。rGREAT 将 DMR 关联至邻近基因,并评估这些基因在 Gene Ontology (GO) 及 KEGG 功能类别中的富集程度,输出区域—基因关联结果与 GO/KEGG 富集结果,为通路解析与机制推断提供依据。
HOMER motif 分析
在单细胞甲基化数据分析中,应将差异甲基化区域 (DMR/VMR) 的序列映射为潜在转录因子 (TF) 结合位点,构建“甲基化变化 → TF 占据 → 基因调控 → 表达差异”的因果链,并将富集的 TF 与其靶基因链接,解析细胞类型/状态转变、疾病驱动与治疗响应中的上游调控因素。相较仅进行 GO/KEGG 富集,motif 分析直接指向调控因子层面,更有助于提出机制性假说与设计验证路径。
HOMER 分析文档:甲基化+RNA双组学-差异甲基化区域Motif富集分析.ipynb
提供基于 HOMER 的分析流程,用于在基因组区域(如 ChIP-seq peaks、DMRs 等)开展 DNA 序列 motif 富集,识别潜在 TF 结合位点,并支持将结果用于上游调控解析与 TF-靶基因关联。
MethylTree 谱系树分析
单细胞 DNA 甲基化模式可反映细胞状态差异和一定的克隆/谱系相关信息。基于不同细胞在 genomic bin 层面的 CpG 甲基化相似性,可以构建细胞间距离关系,用于观察细胞群内部的层级结构、推断潜在克隆结构,并结合 RNA 注释或细胞类型信息解释细胞异质性。
MethylTree 谱系树分析教程:甲基化+RNA双组学-MethylTree谱系树分析.ipynb
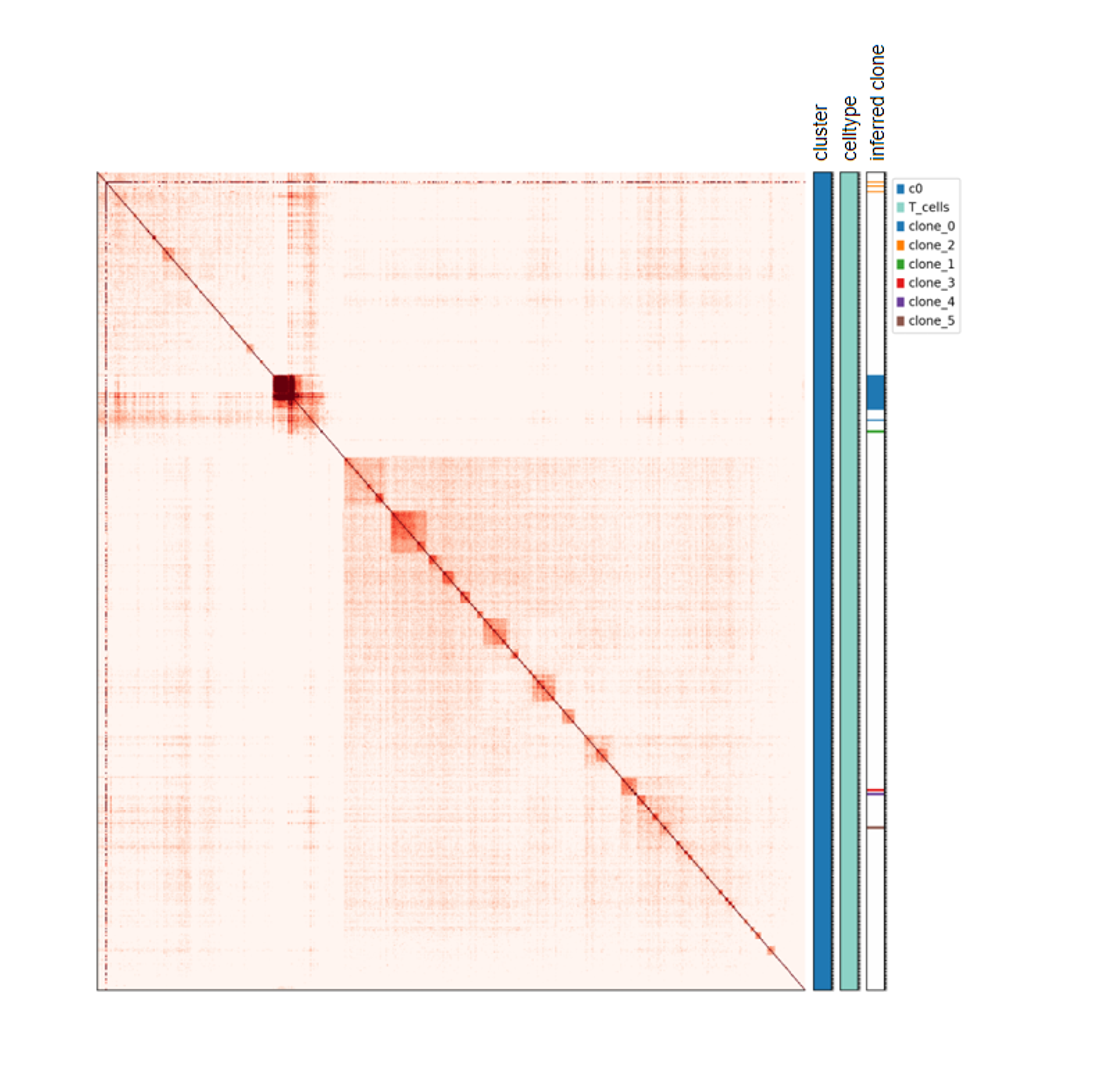
该教程基于单细胞 allc 文件和细胞注释表,先将 CpG 甲基化信号聚合到固定长度的 genomic bin,生成 MethylTree 主程序所需的输入文件;随后运行 MethylTree,完成细胞间相似性计算、克隆结构推断(clone inference)和热图可视化。该分析适用于希望从甲基化层面解析细胞谱系关系、克隆结构或肿瘤异质性的项目。
主要结果包括细胞间甲基化相似性热图、克隆结构推断结果,以及带 inferred clone 注释的可视化图件。
Copykit CNV 分析
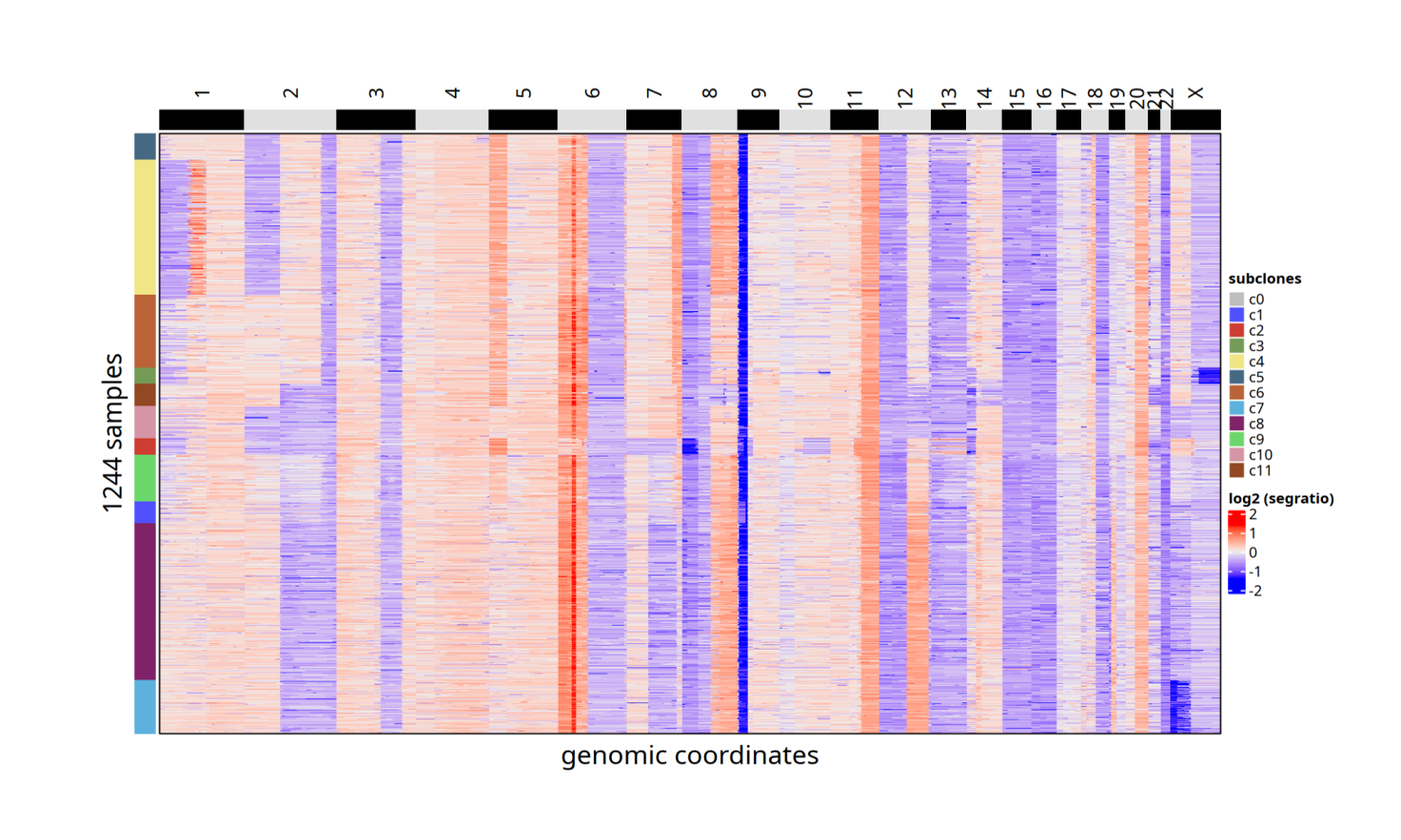
针对肿瘤组织,可使用 Copykit 在单细胞甲基化数据上开展拷贝数变异 (CNV) 分析。Copykit 基于比对得到的 BAM 文件,在基因组预设分箱/区域内统计 reads 深度,关注区域层面的信号而非单核苷酸,从而区分肿瘤与正常细胞,解析肿瘤细胞异质性,构建克隆谱系与进化轨迹,并识别主导与次级亚克隆。
Copykit 肿瘤细胞判定与克隆分析文档:甲基化+RNA双组学-CNV拷贝数变异分析.ipynb
该文档针对三种输入场景(已有 Copykit rds、单细胞 BAM、仅 Bulk BAM)提供数据准备与运行示例,涵盖“BAM 拆分 → flag 校正 → UMI 去重 → runVarbin”关键步骤、命令与参数,帮助用户快速完成肿瘤细胞判定与克隆解析。
数据分析建议
提示:教程基于甲基化双组学数据,旨在为数据整合、差异解析与机制推断提供可操作的参考,具备通用性;但未必完全适用于所有项目分析情况,请结合样本特性与研究目标进行适配与取舍。
建议流程:
- 先完成 “双细胞鉴定” 和 “基础整合分析”。
- 再按目标选择 “差异与功能富集”、“MethSCAn”、“HOMER”、“MethylTree”、“Copykit” 分支。
结果衔接:
DMR→HOMER/rGREAT。DEG/DMG→GO/KEGG。MethylTree→甲基化相似性结构与克隆结构解析。CNV→肿瘤克隆与分群辅助。