Recommendations for Handling Hemoglobin Gene Expression Contamination
This guide summarizes common strategies and practical recommendations for handling hemoglobin gene contamination encountered in single-cell sequencing analysis.
Sources and Impact of Hemoglobin Gene Contamination
In single-cell transcriptome sequencing (scRNA-seq), high expression of hemoglobin genes (such as HBA1, HBA2, HBB, etc.) is common in samples like peripheral blood and bone marrow, mainly originating from red blood cells or their precursors. High expression of hemoglobin genes can lead to downstream analysis biases (such as clustering, differential expression analysis), affecting the accuracy of cell type annotation[^1][^2].
WARNING
Hemoglobin gene contamination may cause non-erythroid cell populations to be incorrectly clustered or annotated, impacting the reliability of biological conclusions[^1].
Mainstream Handling Strategies
Pre-analysis Filtering
During data preprocessing, it is recommended to filter out highly contaminated cells based on the proportion of hemoglobin gene expression (e.g., the proportion of hemoglobin gene transcripts in each cell). For example, Seurat officially recommends removing cells with hemoglobin gene expression above a certain threshold (such as 5%)[^1][^3].Re-clustering After Removing Hemoglobin Genes
Directly remove hemoglobin-related genes from the expression matrix, then re-select highly variable genes and perform clustering analysis again. This helps improve the resolution of non-erythroid cell populations[^2][^4].Flexible Decisions Based on Tissue Type
For tissues like spleen and bone marrow, the presence of erythroid precursor cells has biological significance. It is not recommended to remove them indiscriminately; instead, handle flexibly based on research objectives and tissue characteristics[^2][^5].
NOTE
Specific thresholds and handling strategies can be adjusted according to sample type, research objectives, and downstream analysis needs[^1][^3][^5].
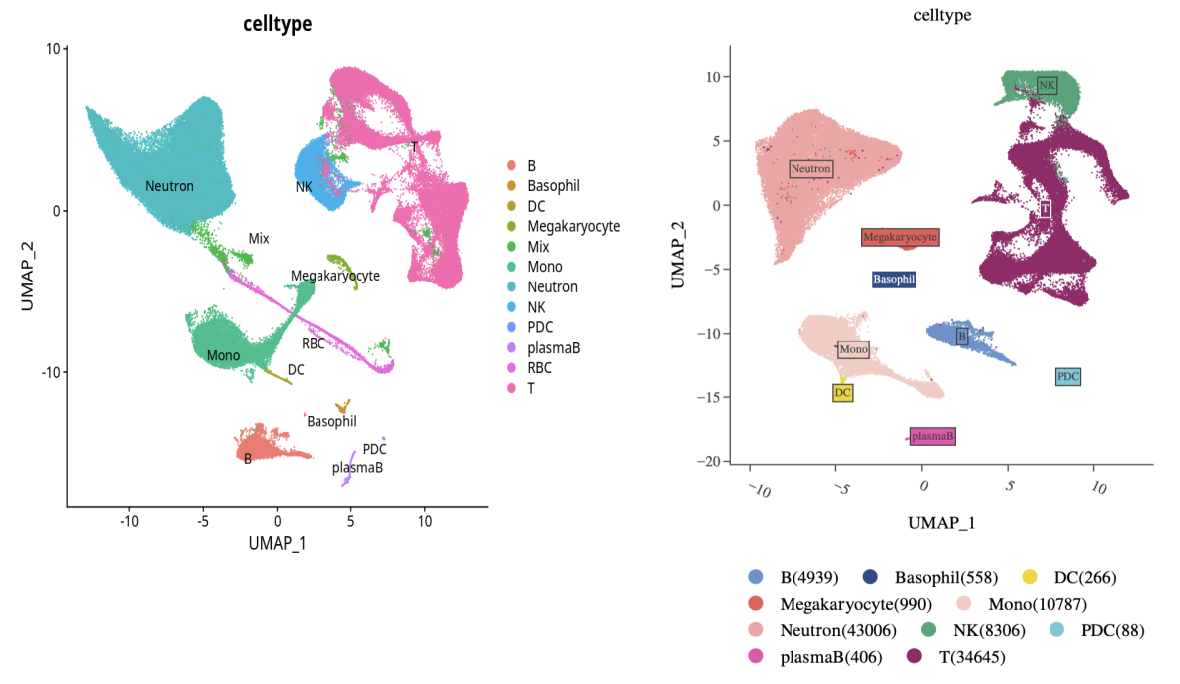
Case 1: Impact of Removing Hemoglobin Genes on Analysis
There is some hemoglobin gene contamination, but erythroid cell expression is obvious, and cell type annotation can still be well distinguished.
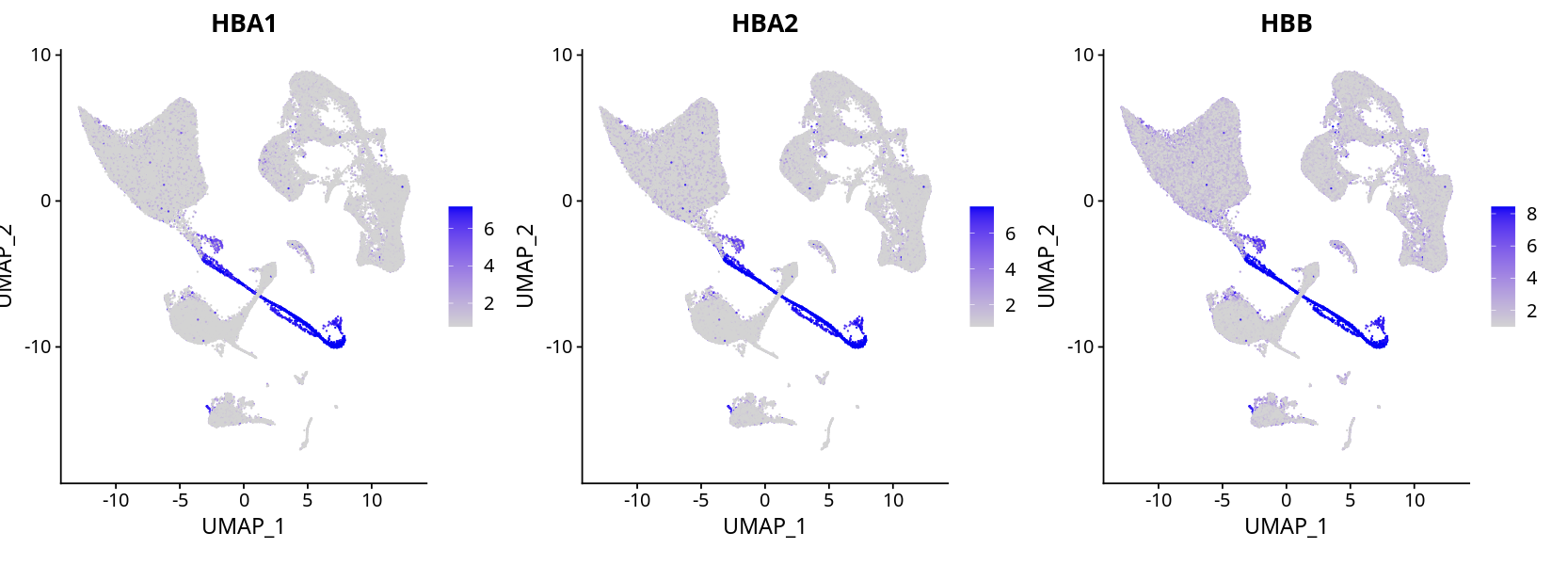
Manually remove mixed cell populations and erythroid cells, and remove hemoglobin genes from the matrix. After re-clustering, cell type distinction is still not a problem.
Left: Original clustering result, Right: Re-clustering after removing hemoglobin genes
IMPORTANT
If erythroid cells are not of interest, hemoglobin genes can be directly discarded, and re-clustering and highly variable gene selection performed.
TIP
For integrated analysis, it is recommended to remove erythroid cells and related genes before downstream analysis.
Reference R code:
counts <- GetAssayData(ob, assay = "RNA")
counts <- counts[-(which(rownames(counts) %in% c("HBA1","HBA2","HBB","HBD","HBE1","HBG1","HBG2","HBM","HBQ1","HBZ"))),]
obj <- subset(ob, features = rownames(counts))
DefaultAssay(obj) <- "RNA"
obj <- FindVariableFeatures(obj, selection.method = "vst", nfeatures = 2000, verbose = FALSE)
obj <- ScaleData(obj, verbose = FALSE)
obj <- RunPCA(obj, npcs = 30, verbose = FALSE)
obj <- FindNeighbors(obj, dims = 1:30)
obj <- FindClusters(obj, resolution = 0.5)
obj <- RunUMAP(obj, reduction = "pca", dims = 1:30)
obj <- RunTSNE(obj, reduction = "pca", dims = 1:30, check_duplicates = FALSE)Case 2: Identification and Handling of Erythroid Precursor Cells
Integrate annotation and check the expression of marker genes for each cell type:
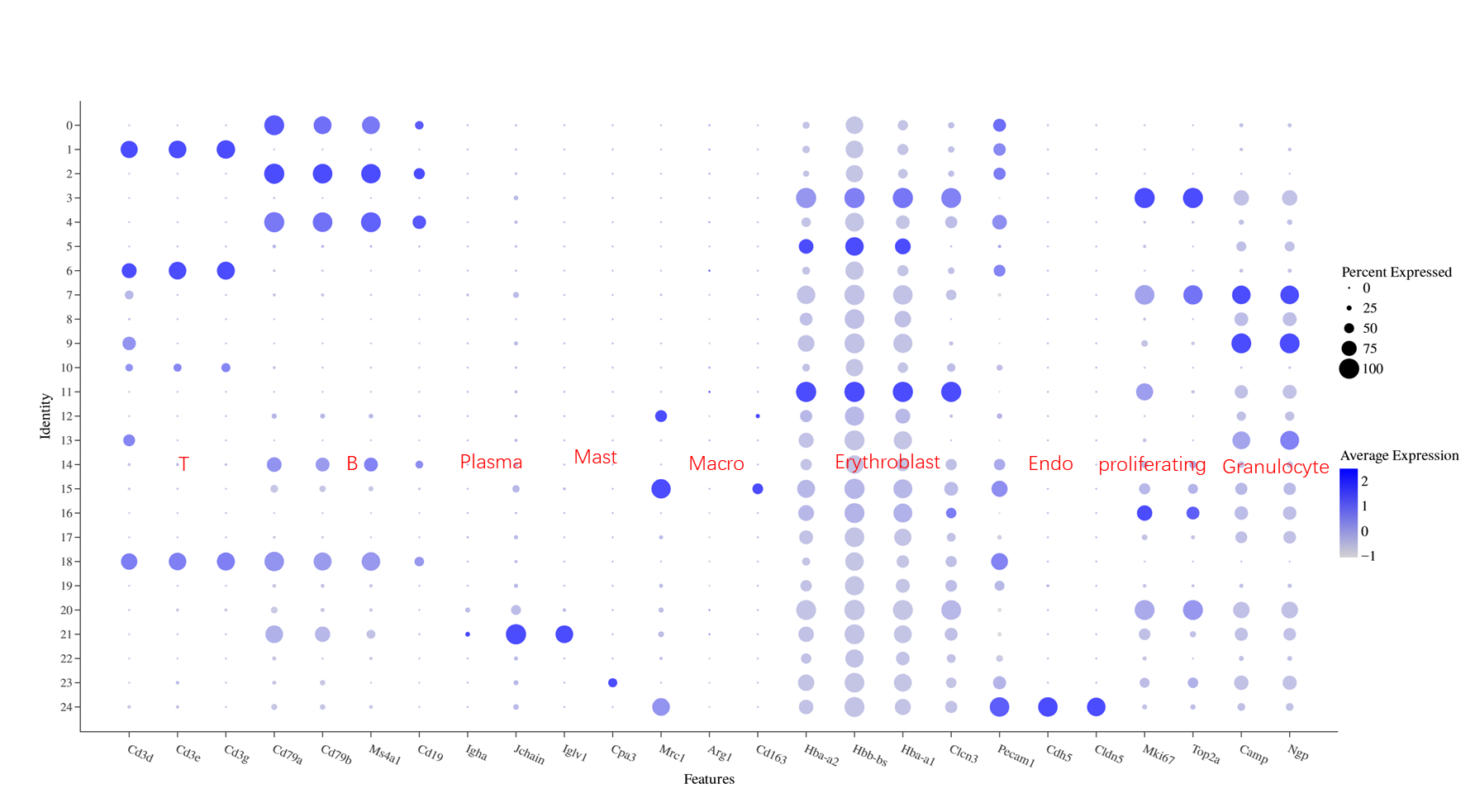
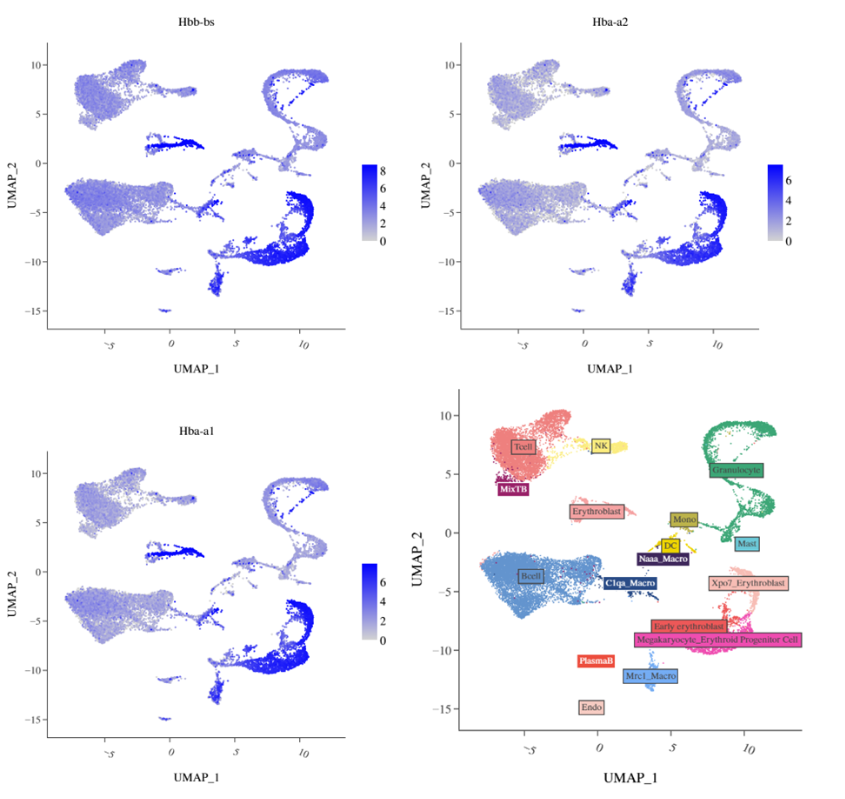
Almost every cluster expresses hemoglobin marker genes, but some clusters (bottom right of UMAP) have higher nFeature and can be specifically annotated as erythroid cells. Annotation of other cell types is not affected. Further, these cell groups express proliferation-related markers.
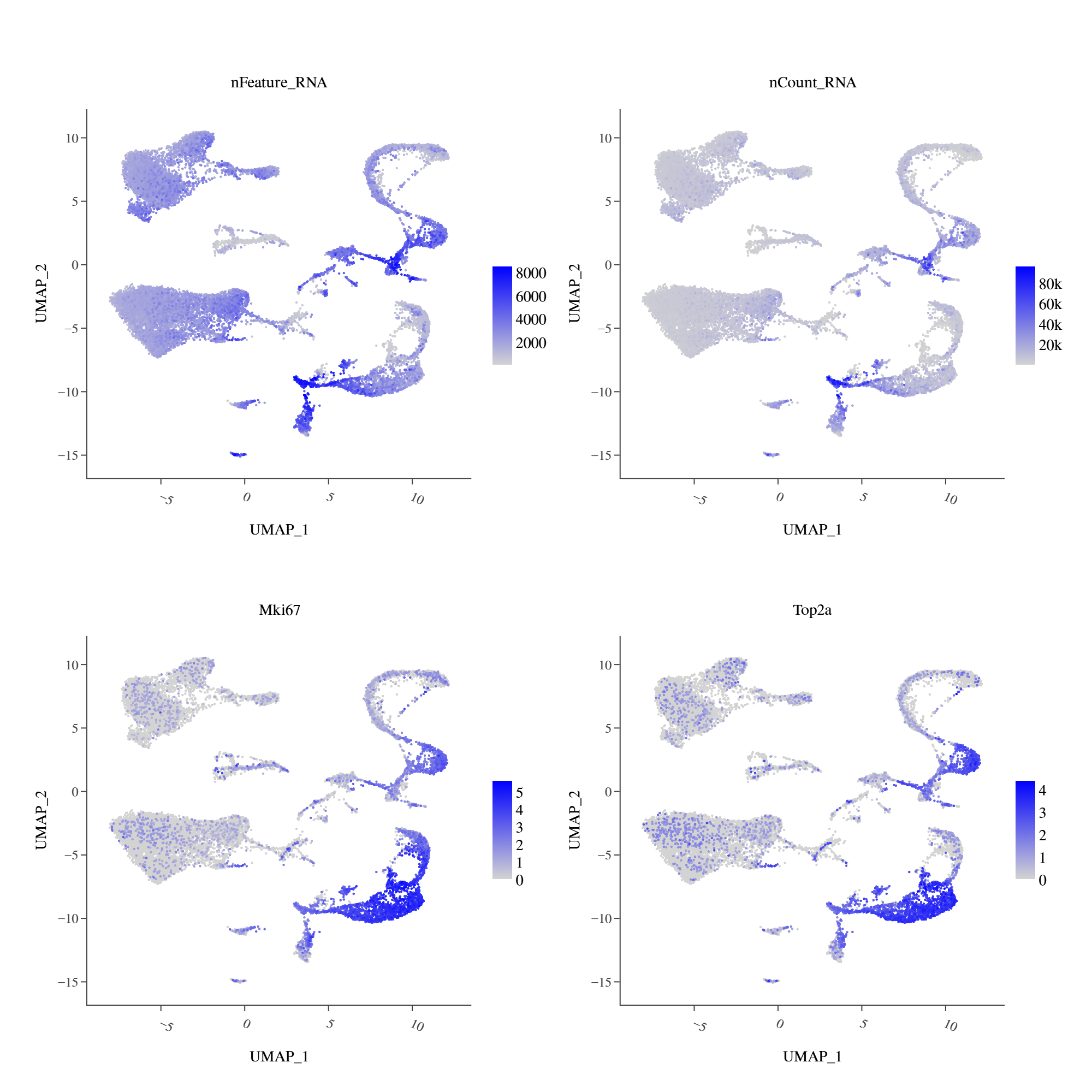
NOTE
These erythroid cells may be erythroid precursor cells. You can further judge based on the specific tissue characteristics of the sample. For example, tissues like spleen and bone marrow may contain cell types related to erythroid development, so it is not recommended to remove them indiscriminately.
TIP
The decision to retain or remove erythroid precursor cells should be made flexibly based on tissue type and research objectives.
Summary and Recommendations
IMPORTANT
- If erythroid cells are not of interest, it is recommended to directly remove hemoglobin-related genes (such as HBA1, HBA2, HBB, etc.) and erythroid cell populations.
- After removal, re-clustering and highly variable gene selection usually do not affect the annotation and analysis of other cell types.
- Some special tissue types (such as spleen, bone marrow, etc.) may contain a large number of erythroid or erythroid precursor cells. It is recommended to make flexible decisions based on research objectives and sample characteristics.
TIP
The processing workflow can be flexibly adjusted. It is recommended to combine actual project needs and downstream analysis goals.
References
[^1]: Hao, Y., Hao, S., Andersen-Nissen, E., et al. (2021). Integrated analysis of multimodal single-cell data. Cell, 184(13), 3573-3587.e29. https://doi.org/10.1016/j.cell.2021.04.048
[^2]: Pijuan-Sala, B., Griffiths, J.A., Guibentif, C., et al. (2019). A single-cell molecular map of mouse gastrulation and early organogenesis. Nature, 566(7745), 490-495. https://doi.org/10.1038/s41586-019-0933-9
[^3]: Stuart, T., Butler, A., Hoffman, P., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell, 177(7), 1888-1902.e21. https://doi.org/10.1016/j.cell.2019.05.031
[^4]: Luecken, M.D., Theis, F.J. (2019). Current best practices in single‐cell RNA‐seq analysis: a tutorial. Molecular Systems Biology, 15(6), e8746. https://doi.org/10.15252/msb.20188746
[^5]: Ziegenhain, C., Vieth, B., Parekh, S., et al. (2017). Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell, 65(4), 631-643.e4. https://doi.org/10.1016/j.molcel.2017.01.023