SeekArc Tools v1.1.0
SeekArc Tools v1.1.0 是寻因生物开发的一套处理单细胞 ATAC 多组学数据的软件,该软件分别对转录组文库和 ATAC 文库进行处理,如 barcode 提取与矫正,比对。对每个 barcode 进行定量,进行联合判定细胞,得到下游分析的细胞表达矩阵,可进行下游聚类和差异分析,计算基因和 Peak 的连接关系。
软件下载
SeekArc Tools v1.1.0
Download-SeekArc Tools - MD5: b7408e3580df1cfb94c0d0a4392f0325
wget 下载方式
mkdir seekarctools_v1.1.0
cd seekarctools_v1.1.0
wget -c -O seekarctools_v1.1.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekarctools/seekarctools_v1.1.0.tar.gz"curl 下载方式
mkdir seekarctools_v1.1.0
cd seekarctools_v1.1.0
curl -C - -o seekarctools_v1.1.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekarctools/seekarctools_v1.1.0.tar.gz"软件安装
安装
# decompress
tar zxf seekarctools_v1.1.0.tar.gz
# install
source ./bin/activate
./bin/conda-unpack
# export path in bashrc
export PATH=`pwd`/bin:$PATH
echo "export PATH=$(pwd)/bin:\$PATH" >> ~/.bashrc确认安装
seekarctools --version使用教程
数据准备
测试数据下载
测试数据 - MD5: 45a48572f5ffd00dd6e6d79e3ad495d4(物种:小鼠)
wget 下载方式
wget -c -O demo_data.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/rawdata/demo_data.tar.gz"
# decompress
tar -zxf demo_data.tar.gzcurl 下载方式
curl -C - -o demo_data.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/rawdata/demo_data.tar.gz"
# decompress
tar -zxf demo_data.tar.gz参考基因组下载
Download-human-reference-GRCh38 - MD5: a9ce266ead3d89bc0aa77cfa646ca309
Download-mouse-reference-GRCm38 - MD5: c4f2244f1d0560d5d3b4c71da06fd08f
wget 下载方式
# human
wget -c -O refdata-arc-GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCh38.tar.gz"
# mouse
wget -c -O refdata-arc-GRCm38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCm38.tar.gz"
# decompress
tar -zxvf refdata-arc-GRCh38.tar.gzcurl 下载方式
# human
curl -C - -o refdata-arc-GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCh38.tar.gz"
# mouse
curl -C - -o refdata-arc-GRCm38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/sgarc_demo/reference/refdata-arc-GRCm38.tar.gz"
# decompress
tar -zxvf refdata-arc-GRCh38.tar.gz参考基因组的构建可以参考[如何构建参考基因组](../1_SeekArc Tools/1_构建参考基因组)
运行 SeekArc Tools v1.1.0
运行示例
示例 1:基本用法
为分析设置必要的配置文件,包括样本数据的路径、参考基因组路径等。使用以下命令运行 SeekArc Tools v1.1.0:
seekarctools arc run \
--rnafq1 /path/to/demo/demo_GE_S1_L001_R1_001.fastq.gz \
--rnafq2 /path/to/demo/demo_GE_S1_L001_R2_001.fastq.gz \
--atacfq1 /path/to/demo/demo_arc_S2_L002_R1_001.fastq.gz \
--atacfq2 /path/to/demo/demo_arc_S2_L002_R2_001.fastq.gz \
--samplename demo \
--outdir /path/to/outdir \
--refpath /path/to/reference/GRCh38 \
--chemistry DD_AG \
--include-introns \
--core 16示例 2:设置阈值重新进行 call peaks 或 call cell
若觉得默认参数下判定出的 peaks 或 cells 不符合需求,可调整参数跳过比对等之前的步骤,重新运行以节省时间
seekarctools arc retry \
--samplename demo \
--outdir /path/to/outdir \
--refpath /path/to/reference/GRCh38 \
--core 16 \
--qvalue 0.01 \
--snapshift 0 \
--extsize 200 \
--min_len 200 \
--min_atac_count 1000 \
--min_gex_count 500NOTE
需注意保持之前运行的文件没有删除或移动。--outdir 为 SeekArc Tools v1.1.0 第一次分析后的目录路径。--min_atac_count 需和 --min_gex_count 一起使用,否则不生效。
参数说明
| 参数 | 参数说明 |
|---|---|
| --rnafq1 | 表达文库 R1 FASTQ 数据路径。 |
| --rnafq2 | 表达文库 R2 FASTQ 数据路径。 |
| --atacfq1 | ATAC 文库 R1 FASTQ 数据路径。 |
| --atacfq2 | ATAC 文库 R2 FASTQ 数据路径。 |
| --samplename | 样本名称。 |
| --chemistry | 试剂类型。可选:DD_AG, DD5_AG。 |
| --outdir | 结果输出目录。默认值:./。 |
| --skip_misB | barcode 不允许碱基错配,默认允许一个碱基错配。 |
| --skip_misL | linker 不允许碱基错配,默认允许一个碱基错配。 |
| --skip_multi | 舍弃能纠错为多个白名单 barcode 的 Reads,默认纠错为比例最高的 barcode。 |
| --skip_len | 在适配器筛选后跳过筛选短读取,将使用短读取。 |
| --core | 分析使用的线程数。 |
| --include-introns | 不启用时,只会选择 exon Reads 用于定量;启用时,intron Reads 也会用于定量。 |
| --refpath | 参考基因组路径。 |
| --star_path | 外部 STAR 软件路径。如果参考基因组中的索引由其他 STAR 构建,请指定该路径。 |
| --qvalue | 峰值检测的最小 FDR(q 值)截止值。默认值:0.001。 |
| --nolambda | 若启用则 MACS3 将使用背景 lambda 作为本地 lambda。这意味着 macs3 不会考虑峰值候选区域的局部偏差。 |
| --snapshift | MACS3 峰值检测的移动幅度。默认值:0。 |
| --extsize | MACS3 峰值检测的拓展幅度。默认值:400。 |
| --min_len | 峰的最小长度。如果无,则设置为 "extsize"。默认值:400。 |
| --broad | 若启用则进行广泛的峰值调用,以 UCSC gappedPeak 格式生成结果,该格式封装了峰值的嵌套结构。 |
| --broad_cutoff | 广泛的峰值调用的阈值。默认值:0.1。 |
| --min_atac_count | 定义细胞条形码峰中 ATAC 转置事件的最小数量(ATAC 计数)。 |
| --min_gex_count | 定义细胞条形码中 UMI 的最小数量(GEX 计数)。 |
| -h,--help | 显示参数说明 |
结果文件说明
以下是输出目录结构:每一行代表一个文件或文件夹,用 "├──" 表示,数字表示重要的输出文件。
./
├── outs
│ ├── filtered_feature_bc_matrix 1
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── filtered_peaks_bc_matrix 2
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix 3
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_peaks_bc_matrix 4
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── demo_A_fragments.tsv.gz 5
│ ├── demo_A_fragments.tsv.gz.tbi 6
│ ├── demo_A_mem_pe_Sort.bam 7
│ ├── demo_A_peaks.bed 8
│ ├── demo_E_SortedByName.bam 9
│ ├── demo.rds 10
│ ├── demo_report.html 11
│ └── demo_summary.csv 12
└── analysis- 转录组细胞稀疏矩阵。
- ATAC 细胞稀疏矩阵。
- 转录组原始稀疏矩阵。
- ATAC 原始稀疏矩阵。
- fragments 文件。
- fragments 文件索引。
- ATAC 的 bam 文件。
- peaks 文件。
- 转录组的 bam 文件。
- rds 格式的样本结果。
- 样本的 html 报告。
- 样本的 csv 格式质控信息。
处理过程
表达文库分析
estep1: barcode/UMI 提取
转录组文库结构:
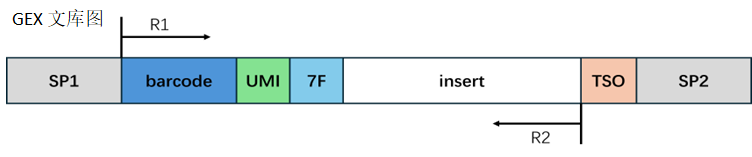
barcode 处理:
根据结构设计,取出相应序列。当取出的 barcode 序列在白名单中时,我们认为它是有效 barcode,计入有效 barcode 的 Reads 数量;当 barcode 不在白名单中时,我们认为它是无效 barcode。
TIP
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekArc Tools v1.1.0 可以尝试 barcode 矫正。在启用矫正时,当无效 barcode 一个碱基错配(一个 hamming distance)的序列存在于白名单中:
- 只有唯一一个序列存在于白名单中:我们将这个无效 barcode 矫正为白名单中 barcode。
- 有多个序列存在于白名单中:我们将这个无效 barcode 改为 Reads 支持数量最多的序列。
接头处理:
在转录组中,Reads 2 的末端有可能会出现 7F 的反向互补序列,建库时可能引入的接头序列。我们对这些污染序列进行切除,剪切完的 Reads 2 长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的 Reads 2 长度小于最小长度,我们认为这条 Reads 为无效 Reads。
estep2: 进行比对并找到比对基因
序列比对
- SeekArc Tools v1.1.0 使用STAR比对软件将处理后的 R2 比对到参考基因组上。
- SeekArc Tools v1.1.0 使用QualiMap软件和转录本注释文件 GTF,统计 Reads 比对外显子、内含子和基因间区等的比例。
- 使用featureCounts将比对上的 Reads 注释到基因上,可以选择不同的注释规则,如链方向性和定量的 feature。当使用外显子定量时,当 Reads 超过 50% 碱基比对到外显子区域时,认为该 Reads 来源于此外显子以及外显子对应的基因;当使用转录本定量时,当 Reads 超过 50% 碱基比对到转录本区域时,认为该 Reads 来源于此转录本以及转录本对应的基因。
经过上述处理后,有如下数据指标:
- Reads Mapped to Genome: 比对到参考基因组上的 Reads 占所有 Reads 的比例(包括只有一个比对位置和多个比对位置的 Reads)。
- Reads Mapped Confidently to Genome: 在参考基因组上只有一个比对位置的 Reads 占所有 Reads 的比例。
- Reads Mapped to Intergenic Regions:比对到基因间隔区 Reads 占所有 Reads 的比例。
- Reads Mapped to Intronic Regions:比对到内含子 Reads 占所有 Reads 的比例。
- Reads Mapped to Exonic Regions:比对到外显子 Reads 占所有 Reads 的比例。
estep3: 定量
UMI 定量:
SeekArc Tools v1.1.0 以 barcode 为单位提取 featureCounts 输出的 bam 数据,统计注释到基因的 UMI 和 UMI 对应的 Reads 数:
- 过滤掉对应 UMI 为单个重复碱基的 Reads,例如 UMI 为 TTTTTTTT。
- 当 Reads 注释到多个基因,在有唯一外显子的注释时,为有效 Reads,其他情形下都过滤掉。
UMI 矫正:
NOTE
测序过程中,UMI 也有一定概几率出现测序错误。SeekArc Tools v1.1.0 默认使用 UMI-tools 的adjacency方法对 UMI 进行矫正。
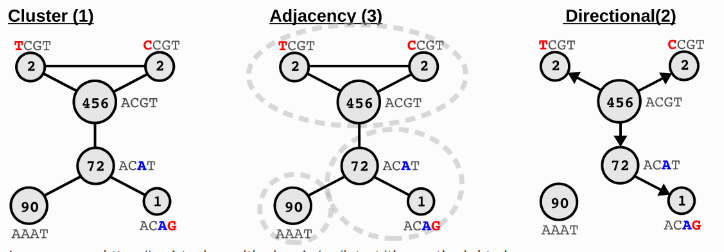
Image source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
ATAC 文库分析
astep1: barcode 提取
ATAC 文库结构:
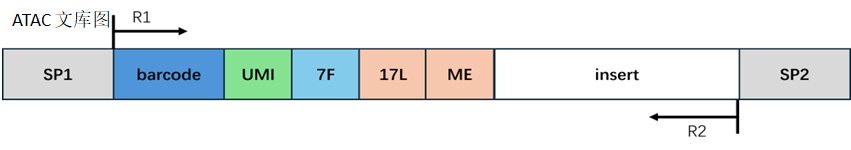
barcode 处理:
同 estep1 的处理方式一样。
接头和 Reads 处理:
在 ATAC 文库中,Reads 1 带有固定序列,末端有可能会出现接头序列;Reads 2 的末端有可能会出现固定序列的反向互补序列和接头序列。我们对这些污染序列进行切除,剪切完的 Reads 1 或 Reads 2 长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的 Reads 1 或 Reads 2 长度小于最小长度,我们认为这条 Reads 为无效 Reads;如果剪切完成后的 Reads 1 或 Reads 2 长度大于 50,则截取 Reads 1 或 Reads 2 序列的前 50bp;如果剪切完成后的 Reads 1 或 Reads 2 长度小于 50 且大于最小长度,则保留 Reads 1 或 Reads 2 的全部序列。
astep2: 进行比对
序列比对:
使用 bwa mem 将处理后的 Reads 1 和 Reads 2 比对到参考基因组上。
astep3: 定量
使用 SnapATAC2 v2.8.0 处理 astep2 输出的 BAM 文件。
- 读取 BAM,获取 ATAC 的 Sequencing 和 Mapping 相关指标。
- 由 BAM 生成原始 fragments 文件。过滤掉线粒体相关的 fragments 后得到过滤后的 fragments 文件。
- 由过滤后的 fragments 文件生成 Anndata 对象。
- 绘制 TSS 得分分布图。
- 使用 MACS3 进行 call peak,并过滤重复的 peaks,输出到 peaks.bed。
- 根据 peaks 生成 raw_peaks_bc_matrix。
- 统计每个 barcode 的相关指标,输出到 per_barcode_metrics.csv。
- 绘制插入片段分布图。
- 细胞判定。
- 生成 filtered_feature_bc_matrix 和 filtered_peaks_bc_matrix。
- 统计 ATAC 的 Cells 和 Targeting 相关指标。
细胞判定
IMPORTANT
若指定 --min_atac_count 和 --min_gex_count:
- 则判定 barcode 中 UMI 数量大于 min_gex_count 值且 events 数量大于 min_atac_count 值的 barcode 为细胞。
若不指定:
- 首先,保留 barcode 中 UMI 数量大于 1 或 events in peaks 数量大于 1 的 barcode。
- 然后,过滤 "the fraction of fragments overlapping called peaks" 小于 "the fraction of genome in peaks" (将 peaks 的范围前后延伸 2kb,如果延伸后的 peak 有重叠则合并两个 peak 的范围)的 barcode。
- 去除重复值。将具有相同 UMI 值和 events in peaks 值的 barcode 过滤。
- 将过滤后的 barcode 的 UMI 值和 events in peaks 值进行 log10 转换。
- 根据转换后的 UMI 值和 events in peaks 值,使用 KMeans 算法将 barcode 分为两群其中均值高的群为细胞,另一群则不是细胞。
- 将去重过滤掉的 barcode 通过判定的 barcode 映射为细胞或非细胞。
- 设置最大细胞数为 20000。
astep4: 后续分析
经过上述步骤,得到细胞的表达矩阵和 ATAC 矩阵后,我们可以进行下一步的分析。
使用 Signac v1.14.0 将细胞的表达矩阵和 ATAC 矩阵合并为一个 seurat object,过滤不在数据库中的 feature,分别对表达矩阵和 ATAC 矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因,进行 WNN 分群,最后计算 feature link。
发布说明
v1.1.0
优化升级
- 添加了
--chemistry参数,可选:DD_AG, DD5_AG。 - 修改 seekarctools_py 为 seekarctools。
- call peak 参数
--qvalue由 0.05 改为 0.001。 - 修改联合判定细胞方法。延伸 peaks 范围。自动判定时,限制最大细胞数为 20000。