SeekSpace® Tools v1.0.0
SeekSpace® Tools 是寻因生物开发的一套处理单细胞空间转录组数据的软件,该软件延用了 SeekSoul Tools 模块进行单细胞转录组的分析,包含 Cell Barcode 定量,判定细胞,得到下游分析的细胞表达矩阵;针对空间文库,SeekSpace® Tools 可以通过芯片上的空间标签的位置和细胞标签的关系将细胞精确定位到芯片上,并且对组织图像中的组织与背景进行分割,对定位成功的细胞进行下游聚类和差异分析,实现对聚类结果的空间可视化。
软件下载
SeekSpace® Tools v1.0.0
Download-SeekSpace® Tools - md5: 8f426fe5d0882870c6c13471bca51f04
wget 下载方式
mkdir seekspacetools_v1.0.0
cd seekspacetools_v1.0.0
wget -c -O seekspacetools_v1.0.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekspacetools/seekspacetools_v1.0.0.tar.gz"curl 下载方式
mkdir seekspacetools_v1.0.0
cd seekspacetools_v1.0.0
curl -C - -o seekspacetools_v1.0.0.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seekspacetools/seekspacetools_v1.0.0.tar.gz"软件安装
安装
# decompress
tar zxf seekspacetools_v1.0.0.tar.gz
# install
source ./bin/activate
./bin/conda-unpack
# export path in bashrc
export PATH=`pwd`/bin:$PATH
echo "export PATH=$(pwd)/bin:\$PATH" >> ~/.bashrc确认安装
seekspacetools --version使用教程
数据准备
测试数据下载
测试数据 - md5: 208efa2d39d5190abec986e9b25aa685(物种:小鼠)
wget 下载方式
wget -c -O demo.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/space/demo.tar"
# decompress
tar xf demo.tarcurl 下载方式
curl -C - -o demo.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/space/demo.tar"
# decompress
tar xf demo.tar参考基因组下载和构建
Download-human-reference-GRCh38 - MD5: 75752934440590827289569e90099408
Download-mouse-reference-GRCm38 - MD5: c784f479d0124238e23547477340b080
wget 下载方式
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
# decompress
tar -zxvf mm10.tar.gzcurl 下载方式
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
# decompress
tar -zxvf mm10.tar.gz参考基因组的构建可以参考如何构建参考基因组
运行
运行示例
示例 1:基本用法
IMPORTANT
为分析设置必要的配置文件,包括样本数据的路径、试剂类型、基因组索引、GTF 等。使用以下命令运行 SeekSpace® Tools:
seekspacetools run \
--fq1 /path/to/demo/demo_expression_S8_L007_R1_001.fastq.gz \
--fq2 /path/to/demo/demo_expression_S8_L007_R2_001.fastq.gz \
--spatialfq1 /path/to/demo/demo_spatial_S7_L007_R1_001.fastq.gz \
--spatialfq2 /path/to/demo/demo_spatial_S7_L007_R2_001.fastq.gz \
--hdmifq /path/to/demo/2P231224030A4.fq.gz \
--samplename demo \
--outdir /path/to/outdir \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDVS \
--core 4 \
--include-introns \
--forceCell 80000 \
--min_umi 200 \
--chip_id 2P231224030A4 \
--DAPI /path/to/demo/2P231224030A4.tifNOTE
--HE 为可选参数,如提供 HE 图片,需要指定 HE 图片路径。
示例 2:支持跳过 Reads 处理步骤,对图片进行调整
seekspacetools run \
--fq1 /path/to/demo/demo_expression_S8_L007_R1_001.fastq.gz \
--fq2 /path/to/demo/demo_expression_S8_L007_R2_001.fastq.gz \
--spatialfq1 /path/to/demo/demo_spatial_S7_L007_R1_001.fastq.gz \
--spatialfq2 /path/to/demo/demo_spatial_S7_L007_R2_001.fastq.gz \
--hdmifq /path/to/demo/2P231224030A4.fq.gz \
--samplename demo \
--outdir /path/to/outdir \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDVS \
--core 4 \
--include-introns \
--forceCell 80000 \
--min_umi 200 \
--chip_id 2P231224030A4 \
--DAPI /path/to/demo/2P231224030A4.tif \
--alignment_file /path/to/demo/parameters.json \
--skipNOTE
--outdir 为 seekspacetools 第一次分析后的目录路径。
软件参数说明
IMPORTANT
| 参数 | 参数说明 |
|---|---|
| --fq1 | 表达文库 R1 FASTQ 数据路径。 |
| --fq2 | 表达文库 R2 FASTQ 数据路径。 |
| --spatialfq1 | 空间文库 R1 FASTQ 数据路径。 |
| --spatialfq2 | 空间文库 R2 FASTQ 数据路径。 |
| --hdmifq | HDMI 文库 FASTQ 数据路径。 |
| --samplename | 样本名称。仅支持数字,字母和下划线。 |
| --outdir | 结果输出目录。默认值:./。 |
| --genomeDir | STAR 构建的参考基因组路径,版本需要与 SeekSpace® Tools 使用的 STAR 一致。 |
| --gtf | 相应物种的 GTF 路径。 |
| --core | 分析使用的线程数。 |
| --chemistry | 试剂类型,每种对应一组 --shift、--pattern、 --structure、--barcode 和 --sc5p 的组合,可选值:DDVS;DDVS 对应 空间转录组试剂盒; |
| --skip_misB | barcode 不允许碱基错配,默认允许一个碱基错配。 |
| --skip_misL | linker 不允许碱基错配,默认允许一个碱基错配。 |
| --skip_multi | 舍弃能纠错为多个白名单 barcode 的 Reads,默认纠错为比例最高的 barcode。 |
| --forceCell | 选用此参数,后面加一个预期的数值 N,SeekSpace® Tools 软件会按照 UMI 从高到低取前 N 个细胞。默认值: 80000。 |
| --min_umi | 细胞的最小 UMI 数,细胞的 UMI 数低于这个数值时会被丢弃。默认值: 200。 |
| --include-introns | 不启用时,只会选择 exon Reads 用于定量;启用时,intron Reads 也会用于定量。 |
| --star_path | 指定其他版本的 STAR 路径进行比对,版本需要与 --genomeDir 版本兼容,默认的 --star_path 为环境下的 STAR。 |
| --chip_id | 芯片 ID。 |
| --DAPI | DAPI 染色的组织图像 (TIFF)。 |
| --HE | H&E 染色的组织图像 (TIFF)。 |
| --alignment_file | 图片调整的参数文件。 |
| --skip | 跳过 Reads 处理步骤,从图片调整处续跑。 |
结果文件说明
以下是输出目录结构:每一行代表一个文件或文件夹,用 "├──" 表示,数字表示重要的输出文件。
./
├── Outs
│ ├── demo_report.html 1
│ ├── demo_summary.csv 2
│ ├── demo_aligned_DAPI.png 3
│ ├── demo_aligned_HE.png (optional) 4
│ ├── demo_aligned_HE_TIMG.png (optional) 5
│ ├── demo_zarr 6
│ ├── demo_filtered_feature_bc_matrix 7
│ │ ├── barcode.tsv.gz
│ │ ├── feature.tsv.gz
│ │ ├── matrix.tsv.gz
│ │ └── cell_location.tsv.gz 8
│ └── clustering
│ └── demo.rds 9
└── Analysis
├── scRNA-seq_Analysis
│ ├── step1
│ ├── step2
│ │ ├── featureCounts
│ │ │ └── demo_SortedByName.bam
│ │ └── STAR
│ │ │ ├── demo_Log.final.out
│ │ │ └── demo_SortedByCoordinate.bam
│ │ └── step3
│ │ ├── filtered_feature_bc_matrix
│ │ └── raw_feature_bc_matrix
├── Spatial_Positioning
│ ├── demo_valid_spatial_umis.csv.gz
│ └── demo_spatial_umis_cleaned.csv.gz
└── Tissue_Detection
└── demo_bc_under_tissue.csvNOTE
- 样本的 html 报告。
- 样本的 csv 格式质控信息。
- 样本的 DAPI 染色图片。
- 样本的 H&E 染色高清图片(可选)。
- 样本的 H&E 染色略缩图(可选)。
- zarr 格式的样本结果。
- 细胞稀疏矩阵。
- 细胞空间坐标文件。
- rds 格式的样本结果。
处理过程
表达文库分析
NOTE
SeekSpace® Tools 使用 SeekSoul Tools rna 模块来进行空间转录组表达文库的分析,具体算法描述见SeekSoul Tools。
空间转录组表达文库的 R1 结构:

细胞判定
IMPORTANT
相较于 SeekSoul Tools,SeekSpace® Tools 在细胞判定过程中默认采用 "forceCell" 方法,默认提取前 80000 个细胞的 UMI 数量,并使用 min_umi 为 200 作为默认阈值,筛选出 UMI 数大于该阈值的细胞作为最终判定的细胞来生成表达矩阵。
空间定位
提取空间标签和位置信息
和空间位置相关的文库有两个,分别是空间文库和 HDMI 文库。
NOTE
空间文库
空间文库的 R1 结构和表达文库一致,R2 结构如下:

对于空间文库 R1,同样使用 SeekSoul Tools 的 rna 模块进行 cell barcode 校正和 UMI 提取。然后,在 R2 上提取 spatial barcode,并生成细胞标签 (cell barcode) 与空间标签 (spatial barcode) 的对应关系。与表达文库的 UMI 所代表的含义不同,空间文库的 UMI (spatial UMI) 代表了每个细胞标签上每个空间标签的表达量。
HDMI 文库 HDMI 文库为单端测序,每条 Reads 包含 32 个碱基的 spatial barcode,每个 spatial barcode 都有相对应的位置信息。我们使用空间文库提取的 spatial barcode 作为白名单,并利用 SeekSoul Tools 的 rna 模块对 HDMI 文库的 spatial barcode 进行校正,同时提取 spatial barcode 对应的空间坐标。
过滤
IMPORTANT
- 在空间文库中提取的 spatial barcode 中,可能会包含一些无效的 barcode。这些无效的 barcode 可能是由于混入了表达文库中长度较短的 mRNA 片段所致。由于这些片段在 HDMI 文库中不存在,因此无法提供相应的位置信息。除了上述提到的因素,测序错误也会产生无效的 barcode。为了确保数据的准确性,我们对这些无效的 spatial barcode 进行了过滤处理,将其排除在分析之外。
- 在 HDMI 文库中,部分 spatial barcode 可能会出现多次,并且每次出现时可能带有不同的位置信息。由于我们无法确定这些 spatial barcode 的确切空间位置,因此这些 spatial barcode 将被过滤掉。
- 针对某些 spatial barcode,我们观察到其对应的 UMI 支持数异常高。我们推测这可能是由于这些 spatial barcode 在实验操作中脱离了芯片,并被液滴包裹所致。这部分 spatial barcode 被认为是不准确的。为了过滤掉这些错误的 spatial barcode,我们采取了以下步骤:
- 将芯片上的位置按照 30x30 的大小划分为多个 bin。
- 统计每个 bin 中的 spatial barcode 的 UMI 支持数。
- 对 bin 按照 UMI 支持数进行降序排序。
- 基于排序后的 bins 的分布,计算阈值。
- 如果某个 bin 中的 UMI 支持数超过阈值,我们将移除该 bin 中 UMI 支持数最多的 cell barcode 对应的所有 spatial barcode。
- 最后,我们将过滤掉所有非细胞的 cell barcode 以及其对应的 spatial barcode,仅对在生物学上具有意义的细胞进行定位。
细胞位置判定
在确定细胞的中心位置时,我们必须考虑一些噪声性的 spatial barcode 的存在。这些 spatial barcode 可能是在实验过程中作为背景存在于液滴中,或者标记在细胞核的碎片上,导致与液滴中其他细胞核上标记为同一细胞标签。这种情况会导致芯片上出现多个中心位置。所以,我们需要对芯片上具有多个中心的细胞进行过滤以确保那些具有明确定义中心位置的细胞被保留。
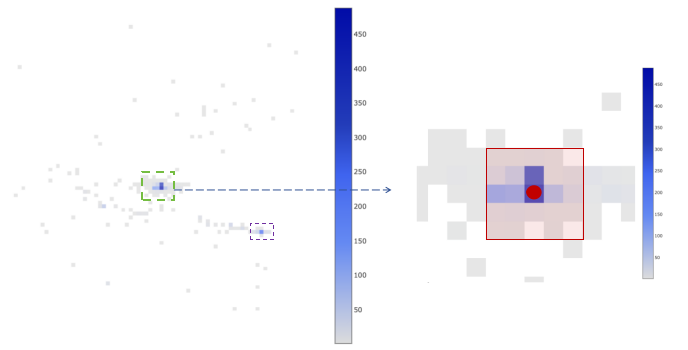
NOTE
在左侧图中,展示了一个细胞的 spatial barcode 在空间中的分布示意图,每个格子代表一个 bin。每个 bin 的大小约为 100 像素,相当于 26.5 微米。图中的颜色表示每个 bin 中 spatial barcode 的 UMI 支持数,颜色越深表示支持数越高。
右侧图是对左侧图中绿色框内的图像进行放大的视角。红点所在的 bin 代表该细胞中 UMI 支持数最高的 bin,被定义为该细胞的中心。红框内包括中心 bin 和周围的 24 个 bin,构成了该细胞的核心。为了判断该细胞是否为多中心,我们需要寻找次中心。次中心被定义为不在核心中的 UMI 支持数最高的 bin。我们计算核心的 UMI 总数与次中心及周围 24 个 bin 中的 UMI 总数之间的比例,如果比例大于等于 2,则认为该细胞具有唯一的中心点。对于其他细胞,我们认为它们具有多个中心点,因此将这些细胞排除。
最后,利用细胞核心中的 spatial barcode 分布,我们可以确定细胞在芯片空间中的位置。
经过上述处理后,有如下数据指标:
IMPORTANT
- Total Spatial Reads: 空间文库中的 Reads 数,表示测序获得的所有 Reads 数量。
- Valid Spatial Reads: 有效的空间 Reads 数,指的是 R1 barcode 不需要校正或校正成功,且 R2 具有至少 32 个碱基长度的 Reads 数量。
- Total Spatial UMIs: 总的空间 UMI 数,表示从有效 Reads 中提取出 cell barcode、 spatial barcode 和 UMI 后进行去重得到的 UMI 数目。
- Spatial barcode Saturation: 空间文库饱和度。1- 总的空间 UMI 数/有效的空间 Reads 数。
- Valid Spatial UMIs: 有效的空间 UMI 数,指的是排除无效 spatial barcode 后剩余的 spatial barcode 的 UMI 支持数。
- Spatial UMIs with Unique Locations: 具有唯一位置的 spatial barcode 的 UMI 支持数占有效的空间 UMI 数的比例。
- Accurate Spatial UMIs: 准确的 spatial barcode 的 UMI 支持数占有效的空间 UMI 数的比例。
- Accurate Spatial UMI Bins: 拥有准确的 spatial barcode 的 bins 占总的 bin 的比例。
- Cell-Identified Spatial UMIs: 与细胞相关的 spatial UMI。被判定为细胞的 cell barcode 上带有的 spatial barcode 的 UMI 支持数占准确的 spatial UMIs 的比例。
- Mean Spatial UMIs per Cell: 每个细胞的平均 spatial UMI 数,与细胞相关的 spatial UMI 数除以判定的细胞数。
组织图像识别
- SeekSpace® Tools 基于图像处理算法识别组织图像并提取组织覆盖下的细胞,以进行后续的分析。它能够接收 DAPI 染色和经过 DAPI 配准后的 HE 染色的图像。输入的图像经过缩放和高斯模糊处理,并利用 OpenCV 中的图像处理算法,最终实现组织和背景的分离。在整个处理过程中,图像的宽高比与原始图像保持一致。
- 如果组织图像和细胞所在区域不完全重合,SeekSpace® Tools 提供了手动对齐功能。通过手动对齐功能,可以对组织图像进行均匀缩放、平移和旋转操作。对齐后,可以下载图片调整参数,并重新输入到 SeekSpace® Tools 中生成新的背景图像,以确保组织图像与细胞区域的准确对应。
后续分析
经过上述步骤,得到被组织覆盖的细胞的表达矩阵后,我们可以进行下一步的分析。
Seurat 分析流程
使用 Seurat 计算线粒体含量,细胞中 UMI 总数,细胞中基因总数。之后对矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因。