SeekSoul Tools v1.2.1
SeekSoulTools 是寻因生物开发的一套处理单细胞转录组数据的软件,目前该软件包含三个模块:
rna模块:用于识别细胞标签barcode,比对定量,得到可用于下游分析的细胞表达矩阵,之后进行细胞聚类和差异分析,该模块不仅支持SeekOne®系列试剂盒产出数据,还可通过对barcode的描述,支持多种自定义设计结构;
fast模块:该模块专门针对SeekOne® DD单细胞全序列转录组试剂盒产出的数据,用于对数据进行barcode提取,双端reads比对,定量,以及全序列数据特有的指标统计。
utils模块:该模块包含其他小工具。
软件下载
SeekSoul Tools v1.2.1
Download-SeekSoulTools - md5: fcd5f0717c8842ee918d8e95881e98fe
wget下载方式
mkdir seeksoultools
cd seeksoultools
wget -c -O seeksoultools.1.2.1.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.1.tar.gz"curl下载方式
mkdir seeksoultools
cd seeksoultools
curl -C - -o seeksoultools.1.2.1.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.1.tar.gz"软件安装
IMPORTANT
安装前请确保系统环境满足要求,并具有足够的磁盘空间。安装完成后必须运行 conda-unpack 和设置环境变量才能正常使用软件。
安装:
# decompress
tar zxf seeksoultools.1.2.1.tar.gz
# install
source ./bin/activate
./bin/conda-unpack
# export path in bashrc
export PATH=`pwd`/bin:$PATH
echo "export PATH=$(pwd)/bin:\$PATH" >> ~/.bashrc确认安装:
seeksoultools --version使用教程
rna模块
数据准备
NOTE
在开始分析之前,请确保已准备好以下必需文件:
- 测序数据(FASTQ格式)
- 对应物种的参考基因组
- 基因注释文件(GTF格式)
测试数据下载
测试数据 - md5: 3d15fcfdefc0722735d726f40ec4e324(物种:人)
wget下载方式
wget -c -O demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tarcurl下载方式
curl -C - -o demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tar参考基因组下载和构建
Download-human-reference-GRCh38 - md5: 5473213ae62ebf35326a85c8fba6cc42
Download-mouse-reference-mm10 - md5: 5c7c63701ffd7bb5e6b2b9c2b650e3c2
wget下载方式
wget -c -O GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gzcurl下载方式
curl -C - -o GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gz参考基因组的构建可以参考如何构建参考基因组?
运行
运行示例
TIP
根据不同的分析需求,SeekSoulTools提供了多种运行模式。以下示例涵盖了最常见的使用场景,可根据实际需要选择合适的参数组合。
示例1:基本用法
为分析设置必要的配置文件,包括样本数据的路径、试剂类型、基因组索引、GTF等。使用以下命令运行SeekSoulTools:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-introns示例2:指定其他版本STAR进行分析
为了在分析中使用特定版本的STAR,并确保与该版本生成的--genomeDir兼容,你可以使用以下命令运行SeekSoulTools,并指定所需版本的STAR的路径:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-introns \
--star_path /path/to/cellranger-5.0.0/lib/bin/STAR示例3:一个样本有多组fastq数据
如果一个样本有多个FASTQ数据集,你可以在运行SeekSoulTools时提供与该样本相关的所有FASTQ文件的路径。以下是一个示例命令:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq1 /path/to/demo_dd_S39_L002_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L002_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-introns示例4:自定义R1结构
要自定义 Read 1 (R1) FASTQ文件的结构,可以使用以下命令:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--barcode /path/to/utils/CLS1.txt \
--barcode /path/to/utils/CLS2.txt \
--barcode /path/to/utils/CLS3.txt \
--linker /path/to/utils/Linker1.txt \
--linker /path/to/utils/Linker2.txt \
--structure B9L12B9L13B9U8 \
--core 4 \
--include-intronsB9L12B9L13B9U8表示read1的结构为:9个碱基barcode+12个碱基linker+9个碱基barcode+13个碱基linker+9个碱基barcode+9碱基UMI,整体cellbarcode有3段,共27个碱基(9*3),UMI为8个碱基- 使用--barcode依次指定3段barcode,--linker依次指定2段linker。
软件参数说明
IMPORTANT
以下参数对分析结果有重要影响,请根据实验设计和数据特点谨慎选择:
- --chemistry:必须与使用的试剂盒类型完全匹配
- --include-introns:影响基因表达定量策略
- --expectNum:影响细胞数量的预估
| 参数 | 参数说明 |
|---|---|
| --fq1 | R1 fastq数据路径。 |
| --fq2 | R2 fastq数据路径。 |
| --samplename | 样本名称,会在outdir目录下创建以样本名称命名的目录。仅支持数字,字母和下划线。 |
| --outdir | 结果输出目录。默认值:./。 |
| --genomeDir | STAR构建的参考基因组路径, 版本需要与SeekSoulTools使用的STAR一致。 |
| --gtf | 相应物种的gtf路径。 |
| --core | 分析使用的线程数。 |
| --chemistry | 试剂类型,每种对应一组--shift、--pattern、 --structure、--barcode和--sc5p的组合,可选值:DDV2,DD5V1,MM,MM-D; DDV2 对应SeekOne(R) DD单细胞3'转录组试剂盒; DD5V1 对应SeekOne(R) DD单细胞5'转录组试剂盒; MM 对应SeekOne(R) MM单细胞转录组试剂盒; MM-D 对应SeekOne(R) MM大孔径高通量转录组试剂盒 |
| --skip_misB | barcode不允许碱基错配,默认允许一个碱基错配。 |
| --skip_misL | linker不允许碱基错配,默认允许一个碱基错配。 |
| --skip_multi | 舍弃能纠错为多个白名单barocde的reads,默认纠错为比例最高的barcode。 |
| --expectNum | 预估的捕获细胞数目。 |
| --forceCell | 当正常分析得到的细胞数⽬不理想时,选⽤此参数,后⾯加⼀个预期的数值N,SeekSoulTools软件会按照UMI从⾼到低取前N个细胞。 |
| --include-introns | 不启用时,只会选择exon reads⽤于定量;启用时,intron reads也会⽤于定量。 |
| --star_path | 指定其他版本的STAR路径进行比对,版本需要与--genomeDir版本兼容,默认的--star_path为环境下的STAR。 |
结果文件说明
以下是输出目录结构:每一行代表一个文件或文件夹,用 "├──" 表示,数字表示三个重要的输出文件。
./
├── demo_report.html 1
├── demo_summary.csv 2
├── demo_summary.json
├── step1
│ └──demo_2.fq.gz
├── step2
│ ├── featureCounts
│ │ └── demo_SortedByName.bam
│ └── STAR
│ ├── demo_Log.final.out
│ └── demo_SortedByCoordinate.bam
├── step3
│ ├── filtered_feature_bc_matrix 3
│ └── raw_feature_bc_matrix
└──step4
├── FeatureScatter.png
├── FindAllMarkers.xls
├── mito_quantile.xls
├── nCount_quantile.xls
├── nFeature_quantile.xls
├── resolution.xls
├── top10_heatmap.png
├── tsne.png
├── tsne_umi.png
├── tsne_umi.xls
├── umap.png
└── VlnPlot.png- 样本的html报告
- 样本的csv格式质控信息
- 算法判定是细胞的稀疏矩阵
处理过程
step1: barcode/UMI提取
CAUTION
barcode和UMI的准确提取对后续分析至关重要。请注意:
- 确保提供正确的结构设计参数
- 注意错配参数的设置
- 监控数据质量指标
SeekSoulTools根据不同的Read1的结构设计和参数对barcode/UMI进行提取和处理,对Read1和Read2进行过滤,输出新的fastq文件。
结构设计和描述
NOTE
以下是Read1结构设计中使用的基本符号说明:
- B: barcode部分碱基
- L: linker部分碱基
- U: UMI部分碱基
- X: 其他任意碱基,用于占位
B: barcode部分碱基 L: linker部分碱基 U: UMI部分碱基 X: 其他任意碱基,用于占位
以下面两种Read1结构为例:B8L8B8L10B8U8: 
B17U12: 
TIP
MM设计下,为了增加Linker部分在测序时碱基均衡性,加入了1-4 bp的移码碱基,即anchor,anchor决定了barcode的起始位置。
数据流程
确定anchor
对于Read1有错位设计的数据(MM试剂产出的数据),seeksoultools尝试在Read1序列前7个碱基中寻找anchor序列,以确定后续barcode等的起始。若没找到anchor序列,此条read以及对应的R2被认为是无效read。
Barcode和Linker矫正
在确定barcode的起始后,根据结构设计,取出相应序列。当取出的barcode序列在白名单中时,我们认为它是有效barcode,计入有效barcode的reads数量;当barcode不在白名单中时,我们认为它是无效barcode。
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekSoulTools可以尝试barcode矫正。在启用矫正时,当无效barcode一个碱基错配(一个hamming distance)的序列存在于白名单中:
- 只有唯一一个序列存在于白名单中:我们将这个无效barcode矫正为白名单中barcode;
- 有多个序列存在于白名单中:我们将这个无效barcode改为read支持数量最多的序列;
Linker的处理与Barcode相同。
接头和PolyA序列剪切
在转录组产品中,Read2的末端有可能会出现polyA tail,建库时可能引入的接头序列。我们对这些污染序列进行切除,剪切完的read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read2长度小于最小长度,我们认为这条read为无效read。
经过上述处理后,数据组成如下图:
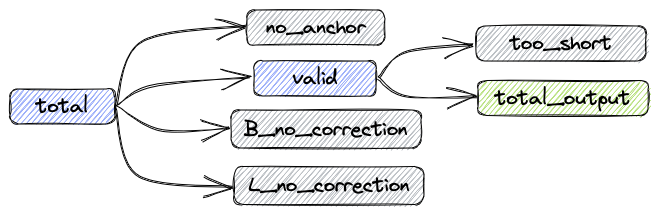
- total: 总共的reads数目
- valid: 不需要矫正和矫正成功的reads数目
- B_corrected: 矫正成功的reads数目
- B_no_correction: 错误Barcode的reads数目
- L_no_correction: 错误Linker的reads数目
- no_anchor:不包含anchor的reads数目
- trimmed: 进行过剪切的reads数目
- too_short: 进行过剪切后长度小于60bp的reads数目
指标之间的关系如下:
total = valid + no_anchor + B_no_correction + L_no_correction
输出fastq的reads数:total_output = valid - too_short
step2: 进行比对并找到比对基因
IMPORTANT
比对质量直接影响下游分析结果。请特别关注:
- STAR版本与参考基因组索引的兼容性
- 比对参数的选择
- 外显子和内含子区域的比对比例
序列比对
- 使用STAR比对软件将处理后的R2比对到参考基因组上。
- 使用QualiMap软件和转录本注释文件GTF,统计reads比对外显子、内含子和基因间区等的比例。
- 使用featureCounts将比对上的read注释到基因上,可以选择不同的注释规则,如链方向性和定量的feature。当使用外显子定量时,当read超过50%碱基比对到外显子区域时,认为该read来源于此外显子以及外显子对应的基因;当使用转录本定量时,当read超过50%碱基比对到转录本区域时,认为该read来源于此转录本以及转录本对应的基因。
NOTE
featureCounts注释规则说明:
- 当使用外显子定量时,read超过50%碱基比对到外显子区域,认为来源于此外显子及其对应基因
- 当使用转录本定量时,read超过50%碱基比对到转录本区域,认为来源于此转录本及其对应基因
经过上述处理后,有如下数据指标:
- Reads Mapped to Genome: 比对到参考基因组上的reads占所有reads的比例(包括只有一个比对位置和多个比对位置的reads)
- Reads Mapped Confidently to Genome: 在参考基因组上只有一个比对位置的reads占所有reads的比例
- Reads Mapped to Intergenic Regions:比对到基因间隔区reads占所有reads的比例
- Reads Mapped to Intronic Regions:比对到内含子reads占所有reads的比例
- Reads Mapped to Exonic Regions:比对到外显子reads占所有reads的比例
step3: 定量
WARNING
定量过程中的关键注意事项:
- UMI去重对表达量估计有重要影响
- 细胞判定的阈值设置需要根据实验设计调整
- 建议仔细检查细胞数量是否符合预期
UMI定量
SeekSoulTools以barcode为单位提取featureCounts输出的bam数据,统计注释到基因的UMI和UMI对应的reads数:
CAUTION
在UMI定量过程中,以下reads会被过滤掉:
- UMI为单个重复碱基的reads(例如UMI为TTTTTTTT)
- 注释到多个基因的reads(除非在有唯一外显子注释的情况下)
UMI矫正
NOTE
测序过程中,UMI也有一定概率出现测序错误。SeekSoulTools默认使用UMI-tools的adjacency方法对UMI进行矫正。
 图片来源: https://umi-tools.readthedocs.io/en/latest/the_methods.html
图片来源: https://umi-tools.readthedocs.io/en/latest/the_methods.html
细胞判定
IMPORTANT
SeekSoulTools使用以下步骤判定barcode是否为细胞:
- 对所有barcode按照对应的UMI数由高到低排序
- 取预估细胞的1%位置的barcode的UMI数除以10为阈值
- barcode的UMI数大于阈值的判定为细胞
- barcode的UMI小于阈值但大于300时,使用DropletUtils分析
- 不符合上述条件的为背景
TIP
DropletUtils分析方法:
- 假设UMI数量低于100的barcode为empty droplet
- 根据每个droplet相同基因的UMI数总和计算背景RNA表达谱
- 通过统计学检验识别显著差异的细胞
经过上述处理后,有如下数据指标:
- Estimated Number of Cells: 算法判定的细胞总数
- Fraction Reads in Cells: 判定为细胞的reads占所有参与定量的reads的比例
- Mean Reads per Cell: 细胞的平均reads数,总reads数/判定的细胞数
- Median Genes per Cell: 判定为细胞的barcode中基因数的中位数
- Median UMI Counts per Cell: 判定为细胞的barcode中UMI数的中位数
- Total Genes Detected: 所有细胞检测到基因数量
- Sequencing Saturation: 饱和度,1 - UMI总数/reads总数
step4: 后续分析
经过上述定量,得到表达矩阵后,我们可以进行下一步的分析。
Seurat分析流程
使用Seurat计算线粒体含量,细胞中UMI总数,细胞中基因总数。之后对矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因。
fast模块
数据准备
测试数据下载
测试数据 - md5: 20b0c7e48cb520d10de5c4b5ee9e0486(物种:人)
wget下载方式
wget -c -O PBMC.tar "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/PBMC.tar"
# decompress
tar xf PBMC.tarcurl下载方式
curl -C - -o PBMC.tar "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/PBMC.tar"
# decompress
tar xf PBMC.tar参考基因组下载和构建
Download-human-reference-GRCh38 - md5: 5473213ae62ebf35326a85c8fba6cc42
Download-hg38-rRNA - md5: 9949f6cea38633daf1d5bf1a2b976488
Download-mouse-reference-mm10 - md5: 5c7c63701ffd7bb5e6b2b9c2b650e3c2
Download-mouse-rRNA - md5: 7a1c2d573086fa9240c8978bb8a859f7
wget下载方式
wget -c -O GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
wget -c -O hg38_rRNA.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/hg38_rRNA.tar.gz"
tar -zxvf hg38_rRNA.tar.gz
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
tar -zxvf mm10.tar.gz
wget -c -O mouse_rRNA.tar.gz "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_rRNA.tar.gz"
tar -zxvf mouse_rRNA.tar.gzcurl下载方式
curl -C - -o GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
curl -C - -o hg38_rRNA.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/hg38_rRNA.tar.gz"
tar -zxvf hg38_rRNA.tar.gz
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
tar -zxvf mm10.tar.gz
curl -C - -o mouse_rRNA.tar.gz "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_rRNA.tar.gz"
tar -zxvf mouse_rRNA.tar.gz参考基因组的构建可以参考如何构建参考基因组?
运行
运行示例
示例1:基本用法
为分析设置必要的配置文件,包括样本数据的路径、试剂类型、基因组索引、GTF等。使用以下命令运行SeekSoulTools:
seeksoultools fast run \
--fq1 /path/to/cellline/cellline_R1.fq.gz \
--fq2 /path/to/cellline/cellline_R2.fq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--rRNAgenomeDir /path/to/hg38_rRNA/star \
--rRNAgtf /path/to/hg38_rRNA/genes/delete_rRNA5.8-18-28_in_rRNA45s.gtf \
--chemistry DD-Q \
--include-introns \
--core 4软件参数说明
| 参数 | 参数说明 |
|---|---|
| --fq1 | R1 fastq数据路径。 |
| --fq2 | R2 fastq数据路径。 |
| --samplename | 样本名称,会在outdir目录下创建以样本名称命名的目录。仅支持数字,字母和下划线。 |
| --outdir | 结果输出目录。默认值:./。 |
| --genomeDir | STAR构建的参考基因组路径, 版本需要与SeekSoulTools使用的STAR一致。 |
| --gtf | 相应物种的gtf路径。 |
| --core | 分析使用的线程数。 |
| --chemistry | 试剂类型,每种对应一组--shift、--pattern、 --structure、--barcode和--sc5p的组合,可选值:DDV2,DD5V1,MM,MM-D; DDV2 对应SeekOne(R) DD单细胞3'转录组试剂盒; DD5V1 对应SeekOne(R) DD单细胞5'转录组试剂盒; MM 对应SeekOne(R) MM单细胞转录组试剂盒; MM-D 对应SeekOne(R) MM大孔径高通量转录组试剂盒 |
| --skip_misB | barcode不允许碱基错配,默认允许一个碱基错配。 |
| --skip_misL | linker不允许碱基错配,默认允许一个碱基错配。 |
| --skip_multi | 舍弃能纠错为多个白名单barocde的reads,默认纠错为比例最高的barcode。 |
| --expectNum | 预估的捕获细胞数目。 |
| --forceCell | 当正常分析得到的细胞数⽬不理想时,选⽤此参数,后⾯加⼀个预期的数值N,SeekSoulTools软件会按照UMI从⾼到低取前N个细胞。 |
| --include-introns | 不启用时,只会选择exon reads⽤于定量;启用时,intron reads也会⽤于定量。 |
| --star_path | 指定其他版本的STAR路径进行比对,版本需要与--genomeDir版本兼容,默认的--star_path为环境下的STAR。 |
结果文件说明
以下是输出目录结构:每一行代表一个文件或文件夹,用 "├──" 表示,数字表示三个重要的输出文件。
.
├── PBMC_report.html
├── PBMC_summary.csv
├── PBMC_summary.json
├── step1
│ ├── PBMC_1.fq.gz
│ ├── PBMC_2.fq.gz
│ ├── PBMC_multi_1.fq.gz
│ ├── PBMC_multi_2.fq.gz
│ └── PBMC_multi.json
├── step2
│ ├── featureCounts
│ │ ├── counts.txt
│ │ ├── counts.txt.summary
│ │ └── PBMC_SortedByName.bam
│ └── STAR
│ ├── downbam
│ │ ├── log.txt
│ │ ├── PBMC.bed
│ │ ├── PBMC.down.0.1.bam
│ │ ├── PBMC.down.0.1.bam.bai
│ │ ├── PBMC.geneBodyCoverage.curves.pdf
│ │ ├── PBMC.geneBodyCoverage.r
│ │ ├── PBMC.geneBodyCoverage.txt
│ │ └── PBMC.reduction.bed
│ ├── PBMC_Log.final.out
│ ├── PBMC_Log.out
│ ├── PBMC_Log.progress.out
│ ├── PBMC_SJ.out.tab
│ ├── PBMC_SortedByCoordinate.bam
│ ├── PBMC_SortedByCoordinate.bam.bai
│ ├── PBMC_SortedByName.bam
│ ├── PBMC__STARtmp
│ ├── report.pdf
│ ├── rnaseq_qc_results.txt
│ └── rRNA
│ ├── counts.txt
│ ├── counts.txt.summary
│ ├── PBMC_Aligned.out.bam
│ ├── PBMC_Aligned.out.bam.featureCounts.bam
│ ├── PBMC_Log.final.out
│ ├── PBMC_Log.out
│ ├── PBMC_Log.progress.out
│ ├── PBMC_SJ.out.tab
│ ├── PBMC__STARtmp
│ └── PBMC.xls
├── step3
│ ├── counts.xls
│ ├── detail.xls
│ ├── filtered_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ └── umi.xls
└── step4
├── biotype_FindAllMarkers.xls
├── FeatureScatter.png
├── FindAllMarkers.xls
├── lncgene_FindAllMarkers.xls
├── mito_quantile.xls
├── nCount_quantile.xls
├── nFeature_quantile.xls
├── PBMC.rds
├── resolution.xls
├── top10_heatmap.png
├── tsne.png
├── tsne_umi.png
├── tsne_umi.xls
├── umap.png
└── VlnPlot.png- PBMC_report.html是样本的html报告
- PBMC_summary.csv样本的csv格式质控信息
- step3/filtered_feature_bc_matrix是经过过滤之后的表达矩阵
- step4/PBMC.rds是矩阵经过seurat处理后的rds文件。
处理过程
step1: barcode/UMI提取
SeekSoulTools根据不同的Read1的结构设计和参数对barcode/UMI进行提取和处理,对Read1和Read2进行过滤,输出新的fastq文件。
结构设计和描述
barcode和UMI描述 以字母和数字描述Read1的基本结构,字母描述碱基含义,数字描述碱基长度。
B: barcode部分碱基 U: UMI部分碱基 X: 其他任意碱基,用于占位
全序列产品的Read1结构为B17U12:
数据流程
Barcode和矫正:
在确定barcode的起始后,根据结构设计,取出相应序列。当取出的barcode序列在白名单中时,我们认为它是有效barcode,计入有效barcode的reads数量;当barcode不在白名单中时,我们认为它是无效barcode。
测序过程中,有一定几率发生测序错误。在提供有白名单的情况下,SeekSoulTools可以尝试barcode矫正。在启用矫正时,当无效barcode一个碱基错配(一个hamming distance)的序列存在于白名单中:
- 只有唯一一个序列存在于白名单中:我们将这个无效barcode矫正为白名单中barcode;
- 有多个序列存在于白名单中:我们将这个无效barcode改为read支持数量最多的序列;
接头和PolyA序列剪切
在建库时可能引入的接头或者TSO等序列,我们对这些污染序列进行切除,剪切完的read1和read2长度需要大于设定的最小长度来保证有足够的信息,准确比对到基因组的位置。如果剪切完成后的read1或read2长度小于最小长度,我们认为这条read为无效read。
经过上述处理后,数据指标包括:
- total: 总共的reads数目
- valid: 不需要矫正和矫正成功的reads数目
- B_corrected: 矫正成功的reads数目
- B_no_correction: 错误Barcode的reads数目
- trimmed: 进行过剪切的reads数目
- too_short: 进行过剪切后长度小于60bp的reads数目
输出fastq的reads数:total_output = valid - too_short
step2: 进行比对并找到比对基因
序列比对
- 使用STAR比对软件将处理后的reads1和reads2提取8Mreads比对到核糖体的参考基因组上,用featureCounts将比对上的read定位到基因上,并统计数据中rRNA和Mt_rRNA比例。
- 使用STAR比对软件将处理后的reads1和reads2比对到参考基因组上。
- 根据gtf文件找到housekeeping基因ACTB的所有exon位置,并从bam中统计覆盖到超过0.2倍ACTB平均深度的区间的比例。
- 根据gtf文件,将全部转录本exon区间转成bed格式,并随机抽出10000个转录本的区间进行genebody绘制。
- 使用QualiMap软件和转录本注释文件GTF,统计reads比对外显子、内含子和基因间区等的比例。
- 使用featureCounts将比对上的read注释到基因上,可以选择不同的注释规则,如链方向性和定量的feature。当使用外显子定量时,当read>超过50%碱基比对到外显子区域时,认为该read来源于此外显子以及外显子对应的基因;当使用转录本定量时,当read超过50%碱基比对到转录本区域时,认为该read来源于此转录本以及转录本对应的基因。
经过上述处理后,有如下数据指标:
- Reads Mapped to Genome: 比对到参考基因组上的reads占所有reads的比例(包括只有一个比对位置和多个比对位置的reads)
- Reads Mapped to Middle Genebody:在genebody中,覆盖转录本的25%-75%区间的比例。
- Reads Mapped Confidently to Genome: 在参考基因组上只有一个比对位置的reads占所有reads的比例
- Fraction over 0.2 mean coverage depth of ACTB gene:超过ACTB基因平均覆盖深度0.2x区间占ACTB基因长度的比例。
- rRNA% in Mapped:数据中核糖体基因的占比。
- Mt_rRNA% in Mapped:数据中线粒体核糖体基因的占比。
- Reads Mapped to Intergenic Regions:比对到基因间隔区reads占所有reads的比例
- Reads Mapped to Intronic Regions:比对到内含子reads占所有reads的比例
- Reads Mapped to Exonic Regions:比对到外显子reads占所有reads的比例
step3: 定量
UMI定量
SeekSoulTools以barcode为单位提取featureCounts输出的bam数据,统计注释到基因的UMI和UMI对应的reads数:
- 过滤掉对应UMI为单个重复碱基的reads, 例如UMI为TTTTTTTT
- 当read注释到多个基因,在有唯一外显子的注释时,为有效read,其他情形下都过滤掉
UMI矫正
测序过程中,UMI也有一定概几率出现测序错误。SeekSoulTools默认使用UMI-tools的adjacency方法对UMI进行矫正。
图片来源: https://umi-tools.readthedocs.io/en/latest/the_methods.html
细胞判定
在一个细胞群中,我们认为细胞和细胞的mRNA的含量不会相差太多。如果一个barcode对应的mRNA的含量很低,我们认为这个barcode的磁珠并没有捕获细胞,mRNA来源于背景。SeekSoulTools会以上面的规则,进行barcode是否为细胞的判定。有以下几个步骤:
- 对所有barcode按照对应的UMI数由高到低排序;
- 取预估细胞的1%位置的barcode的UMI数除以10为阈值;
- barcode的UMI数大于阈值的判定为细胞;
- barocde的UMI小于阈值,但大于300时,使用DropletUtils分析。DropletUtils方法先假设UMI数量低于100的barcode为empty droplet,然后根据每个droplet相同基因的UMI数总和为背景RNA表达谱中该基因UMI数目,进而得到基因UMI数目的期望值。再通过将每个barcode的UMI数进行统计学检验,显著差异的为细胞;
- 不符合上述条件的为背景。
经过上述处理后,有如下数据指标:
- Estimated Number of Cells: 算法判定的细胞总数
- Fraction Reads in Cells: 判定为细胞的reads占所有参与定量的reads的比例
- Mean Reads per Cell: 细胞的平均reads数,总reads数/判定的细胞数
- Median Genes per Cell: 判定为细胞的barcode中基因数的中位数
- Median lnc Genes per Cell:判定为细胞的barcode中biotype属于lncRNA的基因数的中位数
- Median UMI Counts per Cell: 判定为细胞的barcode中UMI数的中位数
- Total Genes Detected: 所有细胞检测到基因数量
- Sequencing Saturation: 饱和度,1 - UMI总数/reads总数
step4: 后续分析
经过上述定量,得到表达矩阵后,我们可以进行下一步的分析。
Seurat分析流程
使用Seurat计算线粒体含量,细胞中UMI总数,细胞中基因总数。之后对矩阵进行归一化、寻找高变基因、降维聚类之后寻找差异基因。
utils模块
addtag
TIP
addtag工具是v1.2.1版本新增的功能,用于:
- 向bam文件添加barcode和umi标签
- 便于后续的可视化和分析
- 提高数据的可追溯性
运行示例
为分析设置必要的配置文件,包括样本的bam文件和step3目录下的umi.xls文件。使用以下命令运行SeekSoulTools:
seeksoultools utils addtag \
--inbam step2/featureCounts/Samplename_SortedByName.bam \
--umifile step3/umi.xls \
--outbam Samplename_addtag.bam软件参数说明
| 参数 | 参数说明 |
|---|---|
| --inbam | 输入的bam,step2 featureCount目录下的bam文件。 |
| --outbam | addtag之后的bam文件名称 |
| --umifile | 输入的umi文件,seeksoultools输出目录step3的umi.xls文件 |
发布说明
v1.2.1
- 更新报告样式
- 增加SP1、SP2和TSO和接头的剪切
- 增加对bam添加addtag的工具
- 增强对非标准gtf的支持
- fast模块支持人和鼠之外的物种
v1.2.0
- 增加去除barcode和umi序列后的read1的fq文件输出
- 添加fast分析模块
- 应用UMI-tools的矫正方法
- 更新read注释到多个基因时的处理规则:在有唯一外显子的注释时,视为有效read
v1.0.0
- 首次发布