全序列-融合模块
介绍:
NOTE
本文档是关于《全序列-融合模块》的使用说明,描述如何获取融合模块的代码、数据,并给出运行示例和结果说明。
融合分析:
IMPORTANT
基因融合是指两个或多个基因在染色体重排过程中发生的结构变化,形成新的融合基因。这里使用STAR-Fusion软件来做分析,该软件首先将RNA-seq reads比对到参考基因组,识别出可能的融合基因对,然后通过进一步的过滤和验证步骤,生成高置信度的融合基因候选列表。通过STAR-Fusion软件识别到的junction-reads,可以根据其上的barcode和UMI标签信息来对应到单细胞上,这些将作为细胞的融合基因标签,进行画图或其他的下游分析。
一、数据获取:
1)参考基因组:
CAUTION
运行STAR-Fusion需要提前构建好基因组,人和小鼠的参考基因组可以在下方获取,其他物种可以参考installing star fusion · STAR-Fusion/STAR-Fusion Wiki来构建。
NOTE
文件较大,分别约30G,请按需下载:
# human
wget -c -O Fusion_refdata_human_GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_refdata_human_GRCh38.tar.gz"
# Decompress
mkdir Fusion_refdata_human_GRCh38
tar -zxvf Fusion_refdata_human_GRCh38.tar.gz -C Fusion_refdata_human_GRCh38# mouse
wget -c -O Fusion_refdata_mouse_mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_refdata_mouse_mm10.tar.gz"
# Decompress
mkdir Fusion_refdata_mouse_mm10
tar -zxvf Fusion_refdata_mouse_mm10.tar.gz -C Fusion_refdata_mouse_mm102)环境:
IMPORTANT
运行环境需包含seeksoultools, STAR-Fusion,及其他所需软件。
# seeksoultools
wget -c -O seeksoultools_fast_mut_dev.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/seeksoultools_fast_mut_dev.tar.gz"
# Decompress
mkdir seeksoultools
tar -zxvf seeksoultools_fast_mut_dev.tar.gz -C seeksoultools
source seeksoultools/bin/activate
seeksoultools/bin/conda-unpack# Fusion_env
wget -c -O Fusion_env.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_env.tar.gz"
# Decompress
mkdir Fusion_env
tar -zxvf Fusion_env.tar.gz -C Fusion_env
source Fusion_env/bin/activate
Fusion_env/bin/conda-unpack3)代码:
TIP
分析代码和测试数据(人类细胞系,BCR-ABL1融合)可以在这里获取:
wget -c -O Fusion_code.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_code.tar.gz"
tar -zxvf Fusion_code.tar.gz二、运行示例:
# Add custom tool directories to PATH
# Note: export seeksoultools first
export PATH="`pwd`/seeksoultools/bin:$PATH"
# making Fusion_env highest priority
export PATH="`pwd`/Fusion_env/bin:$PATH"
bash Fusion_code/Fusion.sh \
-s demo \
-l Fusion_code/testdata/BCR_ABL1_R1.fastq.gz \
-r Fusion_code/testdata/BCR_ABL1_R2.fastq.gz \
-c 8 \
-o ./output \
-g Fusion_refdata_human_GRCh38/ctat_genome_lib_build_dir \
-t Fusion_code/testdata/cellline.rds参数说明:
| parameter | full-name | description |
|---|---|---|
| s | sample | sample name, required |
| l | R1_fq | raw-fastq data for R1; gz compression format, required |
| r | R2_fq | raw-fastq data for R2; gz compression format, required |
| c | CPU | CPU (default: 8) |
| o | outdir | path for output (default: ./output) |
| g | genome_lib_dir | ref-data for STAR-Fusion, required |
| t | rds | Seurat Object, required; meta.data for this object needs to include the "Sample" column |
注:
WARNING
- 本模块仅限于单样本分析,多样本请分别编写脚本并运行
- 输入的R1/R2需为原始全序列/panel测试数据
- 当输入数据是panel数据时,"-t"参数输入的rds,需为对应全序列数据经过单细胞质控过程所得到的rds
- 融合分析需较大资源,测序数据越大,耗时和所需资源就越大,测序100G的数据参考使用32C128GB或更多
三、运行过程
NOTE
原始测序数据经过fastp质控、seeksoultools提取引物后并得到clean数据后,调用STAT-Fusion程序来识别融合事件。根据JunctionReads上的barcode和UMI标签信息,可以分辨哪些细胞是带有融合reads,有多少个UMI是检测到融合reads。将这些作为细胞的新标签,可以进行后续的分析。从中筛选出JunctionReads大于等于10并且细胞数大于等于2的融合。
四、主要结果说明
以下描述主要输出结果,更多输出结果的详细描述可参考STAR-Fusion官方网站 Home · STAR-Fusion/STAR-Fusion Wiki。
STAR/sample/star-fusion.fusion_predictions.tsv

IMPORTANT
该表格包含识别到的所有融合基因结果,详细描述了每个融合的断点、支持reads数目、左右区域信息和PFAM数据库信息等。从LeftBreakpoint和RightBreakpoint可以获得融合断点位置,也是后续区分融合的唯一标识符。支持融合的reads越多,剪接模式符合生物学意义,该结果越可信。
(1)X.FusionName:融合基因
(2)JunctionReadCount:包含在假定融合连接位点处,一条read可以拆分匹配到两侧融合基因的reads数量
(3)SpanningFragCount:包含融合连接的reads数量,该reads的R1端和R2端对应基因不同
(4)est_J:Junction原始读取reads数量(考虑了多重比对和 fusion 转录本的多样性后的估计值)
(5)est_S:Spanning原始读取read counts(考虑了多重比对和fusion转录本的多样性后的估计值)
(6)SpliceType: 融合基因断点是否出现在参考转录本结构注释(例如gencode)提供的参考外显子连接处
(7)LeftGene:融合基因左侧基因
(8)LeftBreakpoint:融合基因断点左侧染色体位置信息(GRCh38)
(9)RightGene:融合基因右侧基因
(10)RightBreakpoint:融合基因断点右侧染色体位置信息(GRCh38)
(11)LargeAnchorSupport:在假定断点两侧是否存在reads的较长碱基序列(≥25)匹配,缺少LargeAnchorSupport的融合基因通常为假阳性
(12)FFPM:支持融合的reads的标准化结果,即每百万总reads数的融合量
(13)LeftBreakDinuc:与断点相邻的左边两个核苷酸。如果左/右接头的结果是GT-AG或GC-AG,那么很可能这是一个拼接断点
(14)LeftBreakEntropy:断点左侧的15个外显子碱基的香农熵(0-2,低熵位点通常被视为可信度较低的断点)
(15)RightBreakDinuc: 与断点相邻的右边两个核苷酸
(16)RightBreakEntropy:断点右侧的15个外显子碱基的香农熵
(17)annots:融合转录本的简单注释output/plots/*.png
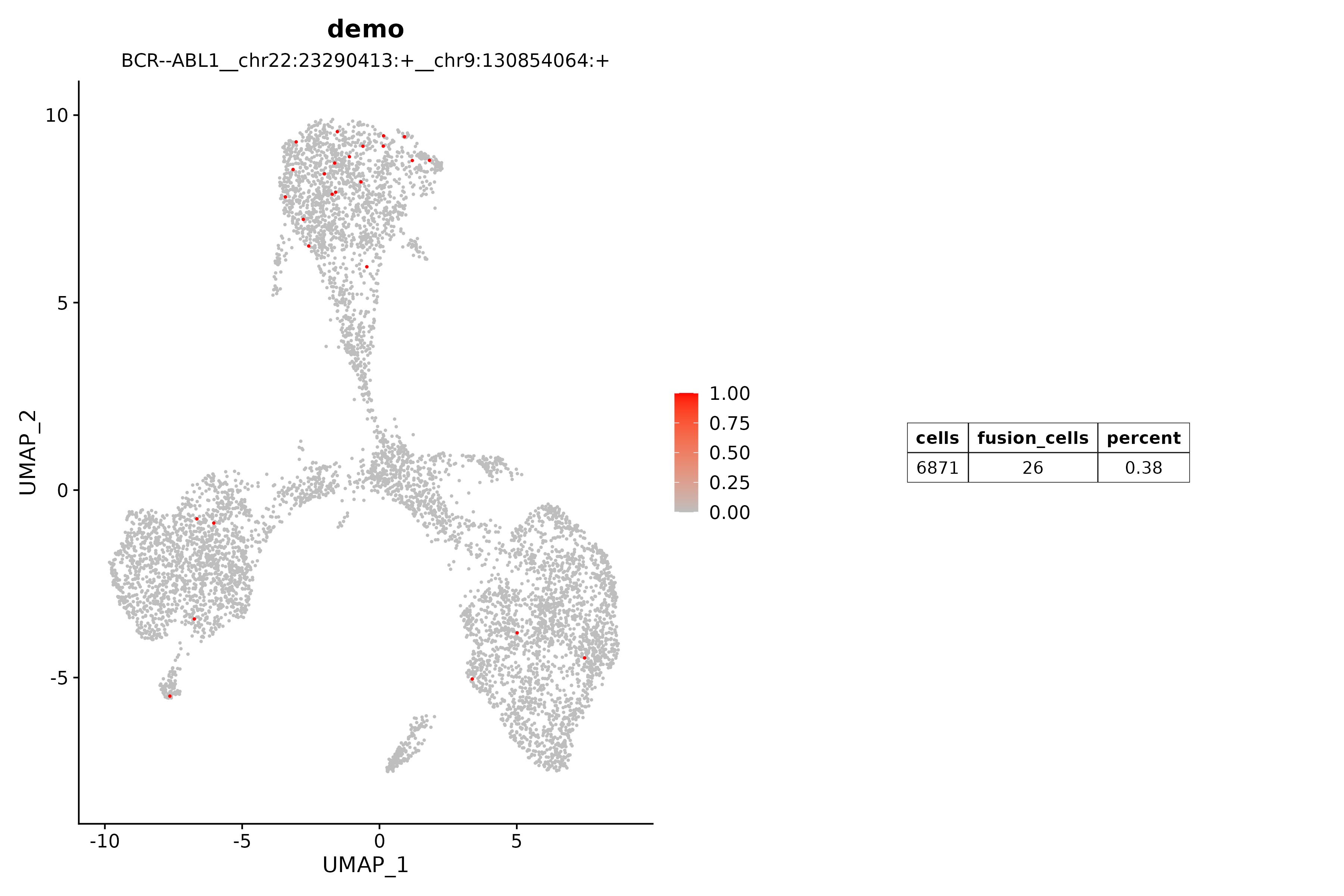
NOTE
对每个融合分别用umap、tsne图展示,图中数值代表检测到该融合的UMI数目,颜色越红代表该细胞表达该融合的值越高。右边表格表示发生该融合的细胞比例,cells表示总细胞数,fusion_cells表示发生该融合的细胞数,percent表示占比。