基于CNV和突变进行肿瘤细胞判定
前言
NOTE
目的:基于突变信息和CNV结果来综合判定肿瘤细胞。
背景:在单细胞转录组数据分析中,识别肿瘤细胞的常规手段包括拷贝数(copy number variation,CNV)推断,标记基因分析和聚类分析等等。文章《Variant calling enhances the identification of cancer cells in single-cell RNA sequencing data》提出用突变和CNV信息来联合分析,不仅可以通过突变和拷贝数变异来分析肿瘤细胞的特征,更重要的是能够从低拷贝数变异的细胞群中识别出肿瘤细胞,从而提供更有价值的结果。

肿瘤细胞判定分析流程
分析思路:
IMPORTANT
- 从inferCNV分析中获得细胞的CNV分数
- 全序列的质控分析获得细胞的突变矩阵,获得突变发生数目和注释信息
- 从OncoKB数据库下载癌症基因表格,用来筛选癌症相关基因
- 全序列的质控分析得到的突变所对应的突变类型包括:"frameshift_variant","inframe_deletion","inframe_insertion","missense_variant","start_lost","stop_gained",即突变类型已满足要求
- 根据突变注释信息表格筛选致病性突变
- 排除HLA基因相关突变
- 分别获取参考细胞和目标细胞的突变数目,CNV结果
- 计算目标细胞的值在正常细胞中的分布位置
- 指定阈值为0.99,满足CNV结果和突变数目任一大于阈值的目标细胞被认为是肿瘤细胞
输入数据:
NOTE
- 样本的alt突变矩阵,突变信息表格(全序列质控分析)
- CNV结果(inferCNV高级分析结果中的CNV_scores.xls表格,要求包含需要分析的细胞,不进行downsample)
- 指定参考细胞,目标细胞
- 指定判断阈值(默认0.99)
这里提供了demo数据,可以直接下载解压,也可以在终端环境中执行命令来获取:
#run in terminal
wget https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/mut_and_cnv_demodata.zip
# decompress
unzip mut_and_cnv_demodata.zip运行过程:
在R环境中输入以下代码:
# run in R
#load packages
library(readr)
library(Seurat)
library(dplyr)
library(ggplot2)
# load data
obj = readRDS("mut_and_cnv_demodata/cellline.rds")
mut_matrix = read.delim("mut_and_cnv_demodata/cellline.snp_indel.alt_UMI.matrix",
header = TRUE,row.names = 1)
mut_info = read.delim("mut_and_cnv_demodata/cellline.anno.cluster.xls",header = TRUE)
CNV_score = read.csv("mut_and_cnv_demodata/cellline_CNV_score.csv")
#specific ref cell and target cell
ref_cell=colnames(obj)[obj$celltype == "ref_cell"]
target_cell=colnames(obj)[obj$celltype == "target_cell"]
#load R code
source("mut_and_cnv_demodata/calling_cancer.R")TIP
运行命令,获取结果:
res=calling_cancer(mut_matrix = mut_matrix,
mut_info = mut_info,
cnv_info = CNV_score,
ref_cells = ref_cell,
target_cells = target_cell,
threshold = 0.99)
p1=callling_cancer_plot(res)
cancer_cell=res$cancer_cell
cancer_label=rep("other",ncol(obj))
cancer_label[match(cancer_cell,colnames(obj))]="cancer_cell"
obj=AddMetaData(obj,metadata=cancer_label,col.name="cancer_label")
p2=DimPlot(obj,group.by="cancer_label",label=T)分析结果:
p1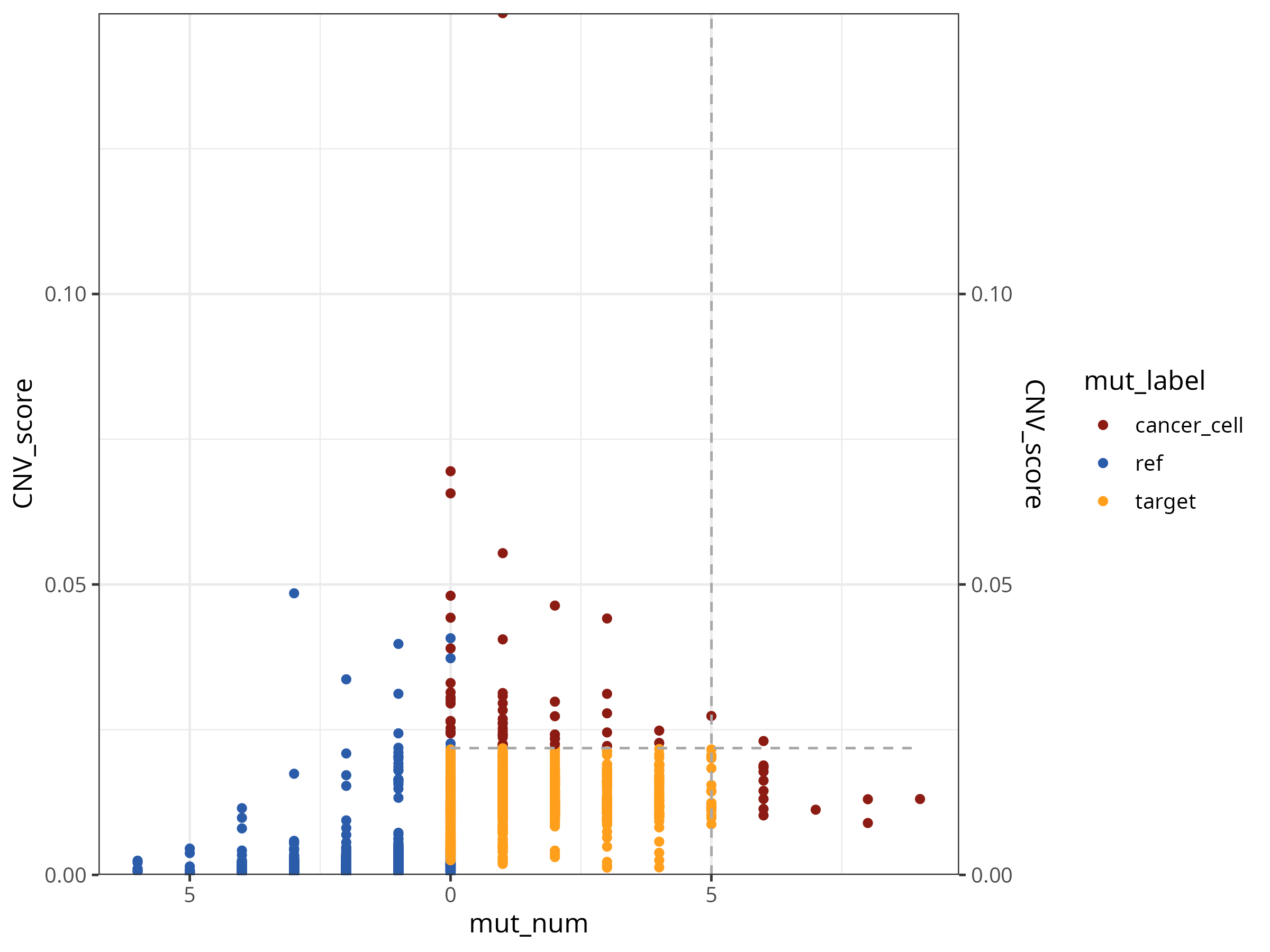
展示参考细胞和目标细胞的突变和CNV信息,并标注最终判定的肿瘤细胞。图中虚线是分别展示了突变和CNV的判定阈值。
p2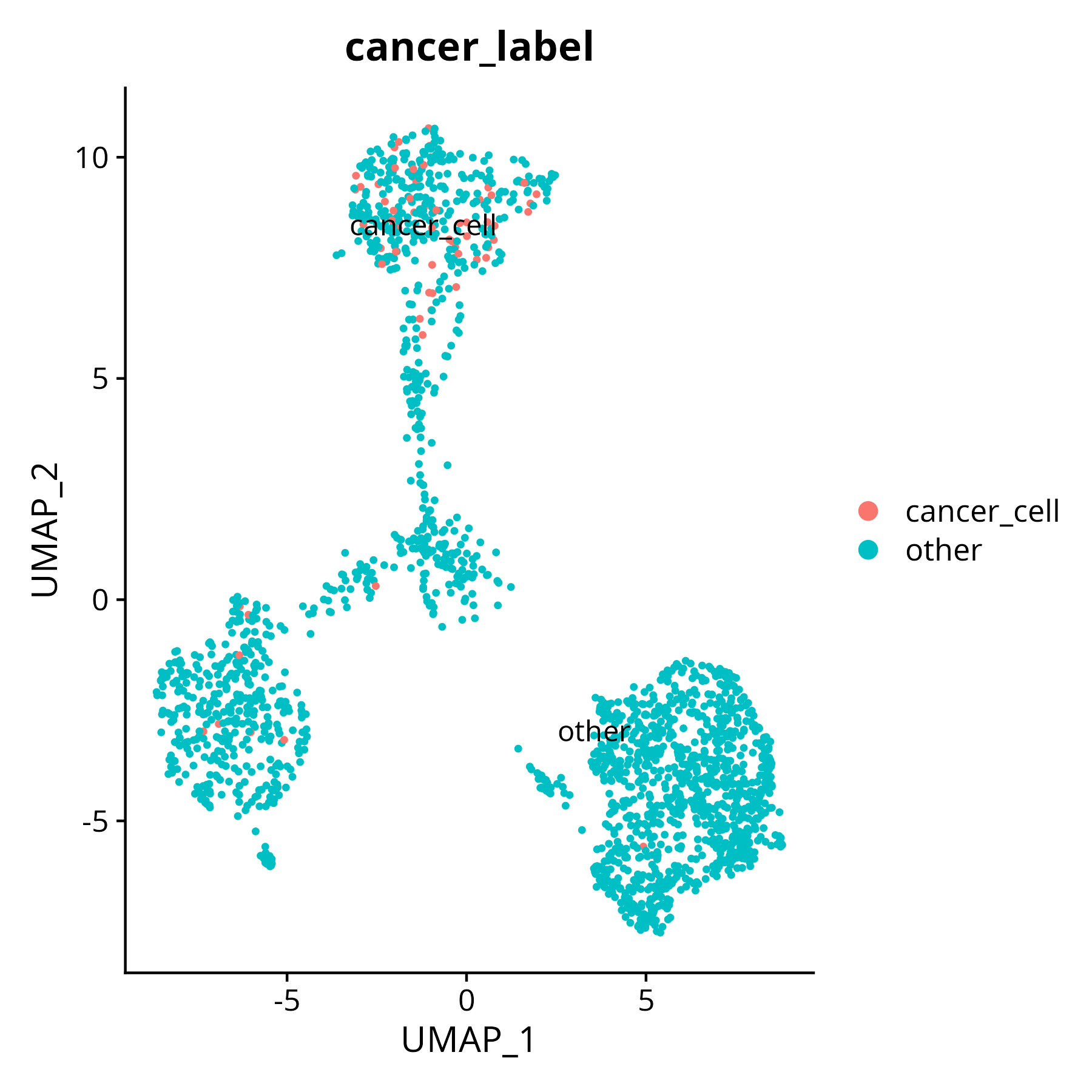
展示将判定的肿瘤细胞作为新的标签添加到object中,可用于后续分析。
保存图片:
ggsave(p1, file = "cellline_calling_cancer.png", width = 8, height = 6)
ggsave(p2, file = "cellline_cancer_label.png", width = 6, height = 6)参考文章:
Gasper W, Rossi F, Ligorio M, Ghersi D (2022) Variant calling enhances the identification of cancer cells in single-cell RNA sequencing data. PLOS Computational Biology 18(10): e1010576. https://doi.org/10.1371/journal.pcbi.1010576
High throughput detection of variation in single-cell whole transcriptome through streamlined scFAST-seq, Guoqin Sang, Jiaxin Chen, Meng Zhao, Huanhuan Shi, Jinhuan Han, Jiacheng Sun, Ying Guan, Xingyong Ma, Guangxin Zhang, Yuyan Gong, Yi Zhao, Shaozhuo Jiao, bioRxiv 2023.03.19.533382; doi: https://doi.org/10.1101/2023.03.19.533382
R包版本:
NOTE
R 4.4.1
readr 2.1.5
Seurat 5.1.0
dplyr 1.1.4
ggplot2 3.5.1