SeekSoul Tools v1.2.1
SeekSoulTools is a software developed by SEEKGENE for processing single-cell transcriptome data. Currently, the software contains three modules:
rna module: This module is used for identifying cell barcodes, genome alignment and gene quantification to obtain a feature-barcode matrix for downstream analysis, followed by cell clustering and differential analysis. This module not only supports data from SeekOne® series kits but also supports various custom structure designs through barcode descriptions.
fast module: This module is specifically designed for data produced by the SeekOne® DD Single Cell Full-length RNA Sequence Transcriptome-seq Kit, used for barcode extraction, paired-end read alignment, quantification, and unique metrics analysis for full-length transcriptome data.
utils module: This module contains additional utility tools.
Software Download
SeekSoul Tools v1.2.1
Download-SeekSoulTools - md5: fcd5f0717c8842ee918d8e95881e98fe
wget download method:
mkdir seeksoultools
cd seeksoultools
wget -c -O seeksoultools.1.2.1.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.1.tar.gz"curl download method:
mkdir seeksoultools
cd seeksoultools
curl -C - -o seeksoultools.1.2.1.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/seeksoultools/seeksoultools.1.2.1.tar.gz"Software Installation
IMPORTANT
Before installation, ensure your system meets the requirements and has sufficient disk space. After installation, you must run conda-unpack and set environment variables for the software to work properly.
Installation:
# decompress
tar zxf seeksoultools.1.2.1.tar.gz
# install
source ./bin/activate
./bin/conda-unpack
# export path in bashrc
export PATH=`pwd`/bin:$PATH
echo "export PATH=$(pwd)/bin:\$PATH" >> ~/.bashrcVerify Installation:
seeksoultools --versionUsage Guide
rna module
Data Preparation
NOTE
Before starting the analysis, ensure you have prepared the following required files:
- Sequencing data (FASTQ format)
- Reference genome for the corresponding species
- Gene annotation file (GTF format)
Download sample datasets
sample datasets - md5: 3d15fcfdefc0722735d726f40ec4e324(Species: Homo sapiens.)
wget
wget -c -O demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tarcurl
curl -C - -o demo_dd.tar "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/demo_dd.tar"
# decompress
tar xf demo_dd.tarDownload and build reference genome
Download-human-reference-GRCh38 - md5: 5473213ae62ebf35326a85c8fba6cc42
Download-mouse-reference-mm10 - md5: 5c7c63701ffd7bb5e6b2b9c2b650e3c2
wget
wget -c -O GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gzcurl
curl -C - -o GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_ensemble_102.tar.gz"
tar -zxvf mm10.tar.gzThe assembly of the reference genome refers to How to build reference genome?
Run SeekSoulTools
Run Examples
TIP
SeekSoulTools provides multiple running modes for different analysis needs. The following examples cover the most common use cases. Choose the appropriate parameter combinations based on your specific requirements.
Example 1: Basic Usage
Set up the necessary configuration files for analysis, including sample data paths, chemistry version, genome index, GTF, etc. Run SeekSoulTools using the following command:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-intronsExample 2: Specify a Different Version of STAR for Analysis
To use a specific version of STAR for analysis while ensuring compatibility with the --genomeDir generated by that version, run SeekSoulTools with the path to the desired STAR version:
seeksoultools rna run \
--fq1 /path/to/demo_dd/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo_dd \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-introns \
--star_path /path/to/cellranger-5.0.0/lib/bin/STARExample 3: Multiple FASTQ Files for One Sample
If a sample has multiple FASTQ datasets, provide the paths to all FASTQ files associated with that sample:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq1 /path/to/demo_dd_S39_L002_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L002_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--chemistry DDV2 \
--core 4 \
--include-intronsExample 4: Custom R1 Structure
To customize the structure of Read 1 (R1) FASTQ files:
seeksoultools rna run \
--fq1 /path/to/demo_dd_S39_L001_R1_001.fastq.gz \
--fq2 /path/to/demo_dd_S39_L001_R2_001.fastq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--barcode /path/to/utils/CLS1.txt \
--barcode /path/to/utils/CLS2.txt \
--barcode /path/to/utils/CLS3.txt \
--linker /path/to/utils/Linker1.txt \
--linker /path/to/utils/Linker2.txt \
--structure B9L12B9L13B9U8 \
--core 4 \
--include-intronsB9L12B9L13B9U8represents the Read1 structure: 9 bases barcode + 12 bases linker + 9 bases barcode + 13 bases linker + 9 bases barcode + 8 bases UMI. The total cell barcode has 3 segments, totaling 27 bases (9*3), and the UMI is 8 bases.- Use --barcode to specify the three barcode segments and --linker to specify the two linker segments sequentially.
Parameter Descriptions
IMPORTANT
The following parameters significantly impact analysis results. Please choose carefully based on your experimental design and data characteristics:
- --chemistry: Must match exactly with the kit type used
- --include-introns: Affects gene expression quantification strategy
- --expectNum: Affects cell number estimation
| Parameters | Descriptions |
|---|---|
| --fq1 | Paths to R1 fastq files. |
| --fq2 | Paths to R2 fastq files. |
| --samplename | Sample name. A directory will be created with this name in the outdir directory. Only digits, letters, and underscores are supported. |
| --outdir | Output directory. Default: ./ |
| --genomeDir | Path to the reference genome generated by STAR. Version must be consistent with the STAR used by SeekSoulTools. |
| --gtf | Path to the GTF file for the corresponding species. |
| --core | Number of threads used for analysis. |
| --chemistry | Reagent type, each corresponding to a combination of --shift, --pattern, --structure, --barcode, and --sc5p. Available options: DDV2, DD5V1, MM, MM-D. DDV2: SeekOne® DD Single Cell 3' Transcriptome-seq Kit DD5V1: SeekOne® DD Single Cell 5' Transcriptome-seq Kit MM: SeekOne® MM Single Cell Transcriptome Kit MM-D: SeekOne® MM Large-well Single Cell Transcriptome-seq Kit |
| --skip_misB | Disallow barcode base mismatches. Default allows one base mismatch. |
| --skip_misL | Disallow linker base mismatches. Default allows one base mismatch. |
| --skip_multi | Discard reads that can be corrected to multiple white-listed barcodes. Default corrects to the barcode with highest frequency. |
| --expectNum | Estimated number of captured cells. |
| --forceCell | When normal analysis yields unsatisfactory cell numbers, use this parameter followed by an expected value N. SeekSoulTools will take the top N cells by UMI count. |
| --include-introns | When disabled, only exon reads are used for quantification; when enabled, intron reads are also used. |
| --star_path | Specify path to alternative STAR version for alignment. Version must be compatible with --genomeDir. Default uses STAR from environment. |
Output descriptions
Here's the output directory structure: each line represents a file or folder, indicated by "├──", and the numbers indicate three important output files.
./
├── demo_report.html 1
├── demo_summary.csv 2
├── demo_summary.json
├── step1
│ └──demo_2.fq.gz
├── step2
│ ├── featureCounts
│ │ └── demo_SortedByName.bam
│ └── STAR
│ ├── demo_Log.final.out
│ └── demo_SortedByCoordinate.bam
├── step3
│ ├── filtered_feature_bc_matrix 3
│ └── raw_feature_bc_matrix
└──step4
├── FeatureScatter.png
├── FindAllMarkers.xls
├── mito_quantile.xls
├── nCount_quantile.xls
├── nFeature_quantile.xls
├── resolution.xls
├── top10_heatmap.png
├── tsne.png
├── tsne_umi.png
├── tsne_umi.xls
├── umap.png
└── VlnPlot.png- Final report in html
- Quality control information in csv
- Filtered feature-barcode matrix
Algorithms Overview
Processing Steps
Step 1: Barcode/UMI Extraction
CAUTION
Accurate extraction of barcodes and UMIs is crucial for downstream analysis. Please note:
- Ensure correct structure design parameters
- Pay attention to mismatch parameter settings
- Monitor data quality metrics
SeekSoulTools extracts and processes barcodes/UMIs according to different Read1 structure designs and parameters, filters Read1 and Read2, and outputs new FASTQ files.
Structure Design and Description
NOTE
Here are the basic symbols used in Read1 structure design:
- B: Barcode bases
- L: Linker bases
- U: UMI bases
- X: Any other bases, used as placeholders
Here are two examples of Read1 structures:
B8L8B8L10B8U8: 
B17U12: 
TIP
In MM design, to increase base balance during sequencing, 1-4 bp shift bases (anchor) are added to the Linker section. The anchor determines the starting position of the barcode.
Data Processing Flow
Determining Anchor Position
For data with shift design (data from MM reagents), SeekSoulTools attempts to find the anchor sequence in the first 7 bases of Read1 to determine the start of subsequent barcodes. If no anchor sequence is found, this read and its corresponding R2 are considered invalid.
Barcode and Linker Correction
After determining the barcode start position, corresponding sequences are extracted based on the structure design. A barcode sequence found in the whitelist is considered a valid barcode and counted in the valid barcode reads count; otherwise, it is an invalid barcode.
NOTE
Sequencing errors can occur during the process. When whitelist is provided, SeekSoulTools can attempt barcode correction. For invalid barcodes with one base mismatch (one hamming distance) to whitelist sequences:
- If only one sequence exists in whitelist: The invalid barcode is corrected to that whitelist barcode
- If multiple sequences exist in whitelist: The invalid barcode is corrected to the sequence with the highest read support
Linker processing follows the same rules as Barcode.
Adapter and PolyA Sequence Trimming
IMPORTANT
In transcriptome products, Read2 may contain:
- PolyA tail at the end
- Adapter sequences from library construction These contaminating sequences are trimmed, and the trimmed read2 must exceed the minimum length for accurate genome alignment.
After the above processing, the data composition is as follows:
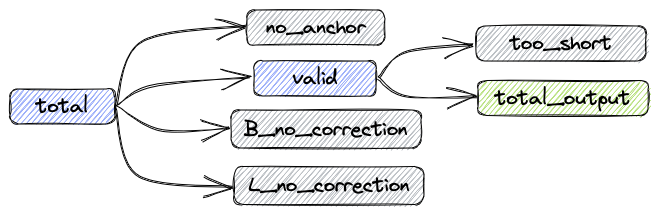
- total: Total number of reads
- valid: Number of reads not requiring correction or successfully corrected
- B_corrected: Number of successfully corrected reads
- B_no_correction: Number of reads with incorrect Barcode
- L_no_correction: Number of reads with incorrect Linker
- no_anchor: Number of reads without anchor
- trimmed: Number of trimmed reads
- too_short: Number of reads shorter than 60bp after trimming
Relationship between metrics:
total = valid + no_anchor + B_no_correction + L_no_correction
Output FASTQ reads count: total_output = valid - too_short
Step 2: Alignment and Gene Assignment
IMPORTANT
Alignment quality directly affects downstream analysis results. Pay special attention to:
- STAR version compatibility with reference genome index
- Alignment parameter selection
- Mapping ratios to exonic and intronic regions
Sequence Alignment
- Uses STAR to align processed R2 to the reference genome.
- Uses QualiMap and GTF file to calculate read distribution across exons, introns, and intergenic regions.
- Uses featureCounts to assign aligned reads to genes, with options for different annotation rules like strand specificity and quantification features.
NOTE
featureCounts annotation rules:
- When using exon quantification: Read is assigned to an exon (and its gene) if >50% bases map to the exon region
- When using transcript quantification: Read is assigned to a transcript (and its gene) if >50% bases map to the transcript region
After processing, the following metrics are available:
- Reads Mapped to Genome: Proportion of reads mapped to reference genome (including unique and multiple mappings)
- Reads Mapped Confidently to Genome: Proportion of reads with unique mapping positions
- Reads Mapped to Intergenic Regions: Proportion of reads mapped to intergenic regions
- Reads Mapped to Intronic Regions: Proportion of reads mapped to intronic regions
- Reads Mapped to Exonic Regions: Proportion of reads mapped to exonic regions
Step 3: Quantification
WARNING
Key considerations during quantification:
- UMI deduplication significantly impacts expression estimation
- Cell calling thresholds should be adjusted based on experimental design
- Carefully check if cell numbers match expectations
UMI Quantification
SeekSoulTools processes featureCounts BAM output by barcode, counting UMIs and corresponding reads assigned to genes:
CAUTION
During UMI quantification, the following reads are filtered out:
- Reads with UMIs consisting of single repeated bases (e.g., TTTTTTTT)
- Reads assigned to multiple genes (except when there's unique exon annotation)
UMI Correction
NOTE
UMIs can also have sequencing errors. SeekSoulTools uses UMI-tools' adjacency method by default for UMI correction.
 Source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Cell Calling
IMPORTANT
SeekSoulTools uses the following steps to determine if a barcode represents a cell:
- Sort all barcodes by UMI count in descending order
- Set threshold as UMI count at 1% of estimated cells divided by 10
- Barcodes with UMI count above threshold are called as cells
- Barcodes with UMI count below threshold but above 300 are analyzed using DropletUtils
- Others are considered background
TIP
DropletUtils analysis method:
- Assumes barcodes with UMI count <100 are empty droplets
- Calculates background RNA expression profile based on total UMIs of same genes
- Identifies significant cells through statistical testing
After processing, the following metrics are available:
- Estimated Number of Cells: Total number of cells determined by algorithm
- Fraction Reads in Cells: Proportion of reads in called cells vs all quantified reads
- Mean Reads per Cell: Average reads per cell (total reads/called cells)
- Median Genes per Cell: Median number of genes in called cell barcodes
- Median UMI Counts per Cell: Median UMI count in called cell barcodes
- Total Genes Detected: Number of genes detected across all cells
- Sequencing Saturation: Saturation level, 1 - (Total UMIs/Total reads)
Step 4: Downstream Analysis
After obtaining the expression matrix through quantification, we can proceed with downstream analysis.
Seurat Analysis Pipeline
Uses Seurat to calculate mitochondrial content, total UMIs per cell, and total genes per cell. Then performs normalization, identifies highly variable genes, dimensionality reduction, clustering, and differential gene analysis.
fast module
Data Preparation
NOTE
Before starting the analysis, ensure you have prepared the following required files:
- Sequencing data (FASTQ format)
- Reference genome for the corresponding species
- Gene annotation file (GTF format)
- rRNA reference files for quality control
Download Sample Datasets
Sample Datasets - md5: 20b0c7e48cb520d10de5c4b5ee9e0486 (Species: Human)
wget download method:
wget -c -O PBMC.tar "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/PBMC.tar"
# decompress
tar xf PBMC.tarcurl download method:
curl -C - -o PBMC.tar "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/demodata/PBMC.tar"
# decompress
tar xf PBMC.tarDownload and Build Reference Genome
Download-human-reference-GRCh38 - md5: 5473213ae62ebf35326a85c8fba6cc42
Download-hg38-rRNA - md5: 9949f6cea38633daf1d5bf1a2b976488
Download-mouse-reference-mm10 - md5: 5c7c63701ffd7bb5e6b2b9c2b650e3c2
Download-mouse-rRNA - md5: 7a1c2d573086fa9240c8978bb8a859f7
wget download method:
wget -c -O GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
wget -c -O hg38_rRNA.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/hg38_rRNA.tar.gz"
tar -zxvf hg38_rRNA.tar.gz
wget -c -O mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
tar -zxvf mm10.tar.gz
wget -c -O mouse_rRNA.tar.gz "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_rRNA.tar.gz"
tar -zxvf mouse_rRNA.tar.gzcurl download method:
curl -C - -o GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/GRCh38.tar.gz"
# decompress
tar -zxvf GRCh38.tar.gz
curl -C - -o hg38_rRNA.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/hg38_rRNA.tar.gz"
tar -zxvf hg38_rRNA.tar.gz
curl -C - -o mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10.tar.gz"
tar -zxvf mm10.tar.gz
curl -C - -o mouse_rRNA.tar.gz "http://seekgene-public.oss-cn-beijing.aliyuncs.com/software/data/reference/mm10_rRNA.tar.gz"
tar -zxvf mouse_rRNA.tar.gzFor reference genome construction, please refer to How to build reference genome?
Running
Run Examples
Example 1: Basic Usage
Set up the necessary configuration files for analysis, including sample data paths, chemistry version, genome index, GTF, etc. Run SeekSoulTools using the following command:
seeksoultools fast run \
--fq1 /path/to/cellline/cellline_R1.fq.gz \
--fq2 /path/to/cellline/cellline_R2.fq.gz \
--samplename demo \
--genomeDir /path/to/GRCh38/star \
--gtf /path/to/GRCh38/genes/genes.gtf \
--rRNAgenomeDir /path/to/hg38_rRNA/star \
--rRNAgtf /path/to/hg38_rRNA/genes/delete_rRNA5.8-18-28_in_rRNA45s.gtf \
--chemistry DD-Q \
--include-introns \
--core 4Parameter Descriptions
IMPORTANT
The following parameters significantly impact analysis results. Please choose carefully based on your experimental design and data characteristics:
- --chemistry: Must match exactly with the kit type used
- --include-introns: Affects gene expression quantification strategy
- --expectNum: Affects cell number estimation
- --rRNAgenomeDir and --rRNAgtf: Required for rRNA content assessment
| Parameters | Descriptions |
|---|---|
| --fq1 | Paths to R1 fastq files. |
| --fq2 | Paths to R2 fastq files. |
| --samplename | Sample name. A directory will be created with this name in the outdir directory. Only digits, letters, and underscores are supported. |
| --outdir | Output directory. Default: ./ |
| --genomeDir | Path to the reference genome generated by STAR. Version must be consistent with the STAR used by SeekSoulTools. |
| --gtf | Path to the GTF file for the corresponding species. |
| --rRNAgenomeDir | Path to the rRNA reference genome index. |
| --rRNAgtf | Path to the rRNA GTF file. |
| --core | Number of threads used for analysis. |
| --chemistry | Reagent type, each corresponding to a combination of --shift, --pattern, --structure, --barcode, and --sc5p. Available options: DDV2, DD5V1, MM, MM-D. DDV2: SeekOne® DD Single Cell 3' Transcriptome-seq Kit DD5V1: SeekOne® DD Single Cell 5' Transcriptome-seq Kit MM: SeekOne® MM Single Cell Transcriptome Kit MM-D: SeekOne® MM Large-well Single Cell Transcriptome-seq Kit |
| --skip_misB | Disallow barcode base mismatches. Default allows one base mismatch. |
| --skip_misL | Disallow linker base mismatches. Default allows one base mismatch. |
| --skip_multi | Discard reads that can be corrected to multiple white-listed barcodes. Default corrects to the barcode with highest frequency. |
| --expectNum | Estimated number of captured cells. |
| --forceCell | When normal analysis yields unsatisfactory cell numbers, use this parameter followed by an expected value N. SeekSoulTools will take the top N cells by UMI count. |
| --include-introns | When disabled, only exon reads are used for quantification; when enabled, intron reads are also used. |
| --star_path | Specify path to alternative STAR version for alignment. Version must be compatible with --genomeDir. Default uses STAR from environment. |
Output Description
Below is the output directory structure. Each line represents a file or folder, indicated by "├──". Numbers indicate important output files:
.
├── PBMC_report.html
├── PBMC_summary.csv
├── PBMC_summary.json
├── step1
│ ├── PBMC_1.fq.gz
│ ├── PBMC_2.fq.gz
│ ├── PBMC_multi_1.fq.gz
│ ├── PBMC_multi_2.fq.gz
│ └── PBMC_multi.json
├── step2
│ ├── featureCounts
│ │ ├── counts.txt
│ │ ├── counts.txt.summary
│ │ └── PBMC_SortedByName.bam
│ └── STAR
│ ├── downbam
│ │ ├── log.txt
│ │ ├── PBMC.bed
│ │ ├── PBMC.down.0.1.bam
│ │ ├── PBMC.down.0.1.bam.bai
│ │ ├── PBMC.geneBodyCoverage.curves.pdf
│ │ ├── PBMC.geneBodyCoverage.r
│ │ ├── PBMC.geneBodyCoverage.txt
│ │ └── PBMC.reduction.bed
│ ├── PBMC_Log.final.out
│ ├── PBMC_Log.out
│ ├── PBMC_Log.progress.out
│ ├── PBMC_SJ.out.tab
│ ├── PBMC_SortedByCoordinate.bam
│ ├── PBMC_SortedByCoordinate.bam.bai
│ ├── PBMC_SortedByName.bam
│ ├── PBMC__STARtmp
│ ├── report.pdf
│ ├── rnaseq_qc_results.txt
│ └── rRNA
│ ├── counts.txt
│ ├── counts.txt.summary
│ ├── PBMC_Aligned.out.bam
│ ├── PBMC_Aligned.out.bam.featureCounts.bam
│ ├── PBMC_Log.final.out
│ ├── PBMC_Log.out
│ ├── PBMC_Log.progress.out
│ ├── PBMC_SJ.out.tab
│ ├── PBMC__STARtmp
│ └── PBMC.xls
├── step3
│ ├── counts.xls
│ ├── detail.xls
│ ├── filtered_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ ├── raw_feature_bc_matrix
│ │ ├── barcodes.tsv.gz
│ │ ├── features.tsv.gz
│ │ └── matrix.mtx.gz
│ └── umi.xls
└── step4
├── biotype_FindAllMarkers.xls
├── FeatureScatter.png
├── FindAllMarkers.xls
├── lncgene_FindAllMarkers.xls
├── mito_quantile.xls
├── nCount_quantile.xls
├── nFeature_quantile.xls
├── PBMC.rds
├── resolution.xls
├── top10_heatmap.png
├── tsne.png
├── tsne_umi.png
├── tsne_umi.xls
├── umap.png
└── VlnPlot.pngKey output files:
PBMC_report.html: Sample HTML reportPBMC_summary.csv: Quality control information in CSV formatstep3/filtered_feature_bc_matrix: Filtered expression matrix after filtering stepsstep4/PBMC.rds: Matrix processed using Seurat
Processing Steps
Step 1: Barcode/UMI Extraction
SeekSoulTools extracts and processes barcodes/UMIs according to different Read1 structure designs and parameters, filters Read1 and Read2, and outputs new FASTQ files.
Structure Design and Description
The basic structure of Read1 is described using letters and numbers, where letters indicate base meanings and numbers indicate base lengths.
B: Barcode bases, U: UMI bases, X: Any other bases (placeholder)
Full-length product Read1 structure B17U12:
Data Processing Flow
CAUTION
During barcode and UMI extraction:
- Monitor extraction quality metrics
- Check for high rates of barcode correction
- Verify trimming statistics
Barcode and Correction:
After determining the barcode start position, corresponding sequences are extracted based on the structure design. A barcode sequence found in the whitelist is considered a valid barcode and counted in the valid barcode reads count; otherwise, it is an invalid barcode.
Sequencing errors can occur during the process. When whitelist is provided, SeekSoulTools can attempt barcode correction. For invalid barcodes with one base mismatch (one hamming distance) to whitelist sequences:
- If only one sequence exists in whitelist: The invalid barcode is corrected to that whitelist barcode
- If multiple sequences exist in whitelist: The invalid barcode is corrected to the sequence with the highest read support
Adapter and TSO Sequence Trimming
IMPORTANT
The following sequences are trimmed:
- Adapter sequences from library construction
- TSO and other technical sequences Trimmed read1 and read2 must exceed minimum length for accurate genome alignment.
After processing, the following metrics are available:
- total: Total number of reads
- valid: Number of reads not requiring correction or successfully corrected
- B_corrected: Number of successfully corrected reads
- B_no_correction: Number of reads with incorrect Barcode
- trimmed: Number of trimmed reads
- too_short: Number of reads shorter than 60bp after trimming
Output FASTQ reads count: total_output = valid - too_short
Step 2: Alignment and Gene Assignment
Sequence Alignment
IMPORTANT
The alignment process includes:
- rRNA content evaluation using 8M reads
- Full dataset alignment to reference genome
- Gene body coverage analysis
- Read distribution analysis
- Uses STAR to align 8M reads from reads1 and reads2 to rRNA reference genome
- Uses featureCounts to assign aligned reads to genes and calculate rRNA and Mt_rRNA proportions
- Uses STAR to align all reads to reference genome
- Analyzes ACTB gene coverage and gene body coverage
- Uses QualiMap and GTF to calculate read distribution across genomic features
- Uses featureCounts for gene assignment with configurable annotation rules
After processing, the following metrics are available:
- Reads Mapped to Genome: Proportion of reads mapped to reference genome
- Reads Mapped to Middle Genebody: Proportion covering 25%-75% of transcript regions
- Reads Mapped Confidently to Genome: Proportion of uniquely mapped reads
- Fraction over 0.2 mean coverage depth of ACTB gene: Proportion of ACTB gene regions with >0.2x mean coverage
- rRNA% in Mapped: Proportion of ribosomal RNA reads
- Mt_rRNA% in Mapped: Proportion of mitochondrial ribosomal RNA reads
- Reads Mapped to Intergenic/Intronic/Exonic Regions: Read distribution across genomic features
Step 3: Quantification
UMI Quantification
WARNING
During UMI quantification:
- Monitor UMI quality metrics
- Check cell number estimates
- Verify saturation levels
SeekSoulTools processes featureCounts BAM output by barcode, counting UMIs and corresponding reads assigned to genes:
- Filters out reads with single-base UMIs (e.g., TTTTTTTT)
- Handles multi-gene assignments based on unique exon annotation
UMI Correction
NOTE
UMIs can have sequencing errors. SeekSoulTools uses UMI-tools' adjacency method by default for UMI correction.
Source: https://umi-tools.readthedocs.io/en/latest/the_methods.html
Cell Calling
IMPORTANT
Cell calling process:
- Sort barcodes by UMI count
- Calculate threshold from estimated cells
- Apply DropletUtils analysis when needed
- Verify final cell counts match expectations
After processing, the following metrics are available:
- Estimated Number of Cells: Total cells identified
- Fraction Reads in Cells: Proportion of reads in called cells
- Mean Reads per Cell: Average reads per cell
- Median Genes per Cell: Median genes per cell
- Median lnc Genes per Cell: Median lncRNA genes per cell
- Median UMI Counts per Cell: Median UMIs per cell
- Total Genes Detected: Total genes across all cells
- Sequencing Saturation: 1 - (Total UMIs/Total reads)
Step 4: Downstream Analysis
After obtaining the expression matrix through quantification, we proceed with downstream analysis.
Seurat Analysis Pipeline
Uses Seurat to calculate mitochondrial content, total UMIs per cell, and total genes per cell. Then performs normalization, identifies highly variable genes, dimensionality reduction, clustering, and differential gene analysis.
utils module
addtag
TIP
The addtag tool is a new feature in v1.2.1, used for:
- Adding barcode and UMI tags to BAM files
- Facilitating visualization and analysis
- Improving data traceability
Run Example
Set up the necessary configuration files, including the sample's BAM file and the umi.xls file in the step3 directory. Run SeekSoulTools using the following command:
seeksoultools utils addtag \
--inbam step2/featureCounts/Samplename_SortedByName.bam \
--umifile step3/umi.xls \
--outbam Samplename_addtag.bamParameter Descriptions
| Parameters | Descriptions |
|---|---|
| --inbam | Input BAM file from step2/featureCount directory. |
| --outbam | Output BAM file path with added tags. |
| --umifile | Input UMI file path (step3/umi.xls). |
Release Notes
v1.2.1
- Updated report style
- Added SP1, SP2, and TSO adapter trimming
- Added addtag tool for BAM files
- Enhanced support for non-standard GTF files
- Fast module now supports species beyond human and mouse
v1.2.0
- Added output of Read1 FASTQ file after removing barcode and UMI sequences
- Added fast analysis module
- Implemented UMI-tools correction method
- Updated multi-gene read assignment rules: valid when unique exon annotation exists
v1.0.0
- Initial release