FAST Alternative Splicing Analysis
Introduction
NOTE
Alternative splicing, refers to the process in eukaryotic gene transcription where precursor RNA undergoes different splicing forms to produce multiple different transcripts. During this splicing process, different exons and introns can be selectively removed or retained.
IMPORTANT
SeekGene's FAST technology (scFAST-seq) is based on random primers to capture RNA molecules, achieving full-length coverage at the single-cell transcriptome level, thus creating opportunities and possibilities for alternative splicing analysis. This transcriptome sequencing technology is a high-throughput sequencing technology based on droplet-based principles. Therefore, we recommend performing alternative splicing analysis at the cell population or sample level.
rMATS Analysis Software
I. Software Introduction
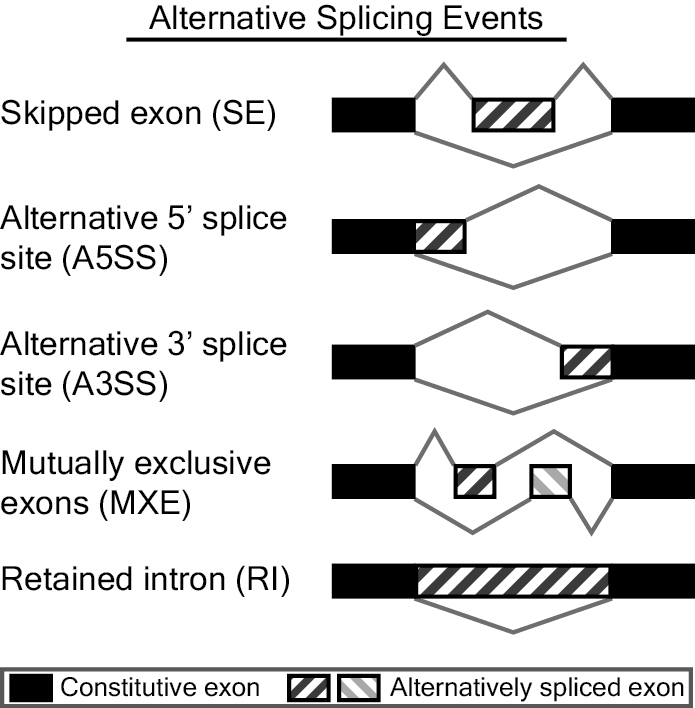
We uses rMATS for alternative splicing analysis. rMATS (Replicate Multivariate Analysis of Transcript Splicing) is a software for analyzing differential alternative splicing events based on RNA-seq data. rMATS can identify different types of alternative splicing events, including five types: SE, A5SS, A3SS, MXE, and RI.
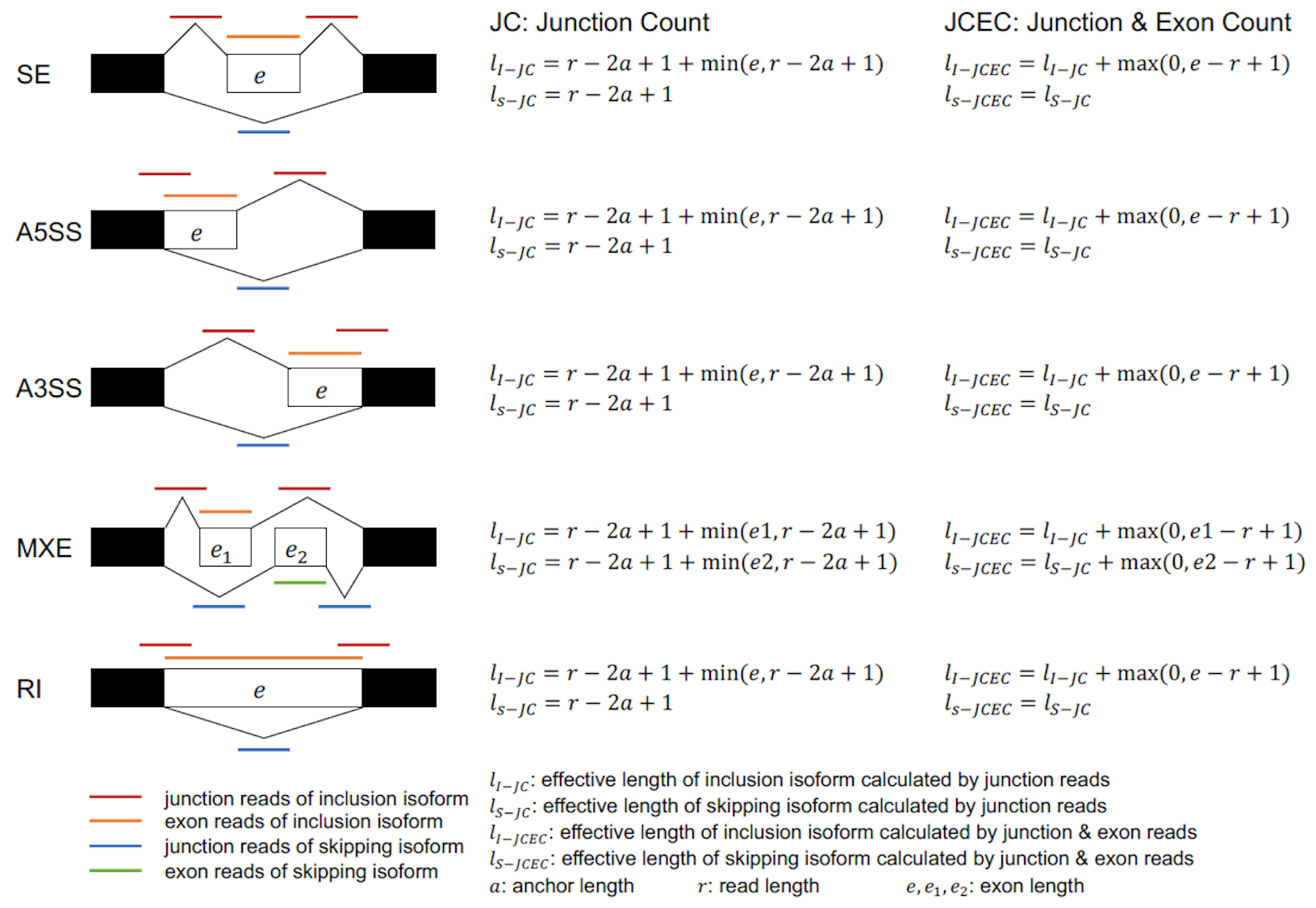
For identified alternative splicing events, rMATS counts supporting reads using two different calculation methods (JC, Junction Counts; JCEC, Junction Counts and Exon Counts).
II. Software Installation
1) Install rMATS using conda:
TIP
We recommends using conda to install the rMATS software. First, create a new conda environment, or you can choose to activate an existing environment:
conda create -n rMATS_env
conda activate rMATS_envInstallation:
conda install bioconda::rmatsAfter successful execution, rMATS is installed.
2) Environment Configuration:
IMPORTANT
To run subsequent code, you need to configure some necessary tools. Install Python packages and R packages according to the following example:
# install python packages
python -m pip install pysam click
# install R
conda install r-base
# install R packages
Rscript -e 'install.packages(c("argparse", "Seurat"))''Example Data and Script Download
Here we provide corresponding example data and analysis scripts to help you understand the subsequent analysis pipeline. The data is sourced from SeekGene's public PBMC data.
wget https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/rMATS_demo.tar.gz
tar -zxvf rMATS_demo.tar.gzNOTE
The example data contains 3 files:
PBMC.rdsis single-cell data in SingleCellExperiment formatPBMC_chr1.bamis alignment data extracted from chromosome 1 of the PBMC sample sequencing data aligned to the GRCh38 genomegenes.gtfis the genome annotation file based on the GRCh38 version
Among the three scripts, getbarcode.R is used to read cell barcode information and grouping information from the rds file; rmats_run.py first splits the bam file according to specified grouping information, then uses rMATS to identify alternative splicing events and calculates differential expression levels between specified groups. run.sh is the direct execution script.
Running Example
CAUTION
Run the following commands to complete alternative splicing analysis:
conda activate rMATS_env# create output path
mkdir -p /path/result
# run
python /path/rmats_run.py \
--samplename demo \
--rds /path/demodata/PBMC.rds \
--outdir /path/result \
--bam /path/demodata/PBMC_chr1.bam \
--gtf /path/demodata/genes.gtf \
--cellanno seurat_clustersThe meanings of the above parameters are:
| Parameter Name | Parameter Meaning |
|---|---|
| samplename | Sample name |
| rds | Data in SingleCellExperiment format, Seurat object |
| outdir | Output directory |
| bam | Alignment result file |
| gtf | Genome annotation file |
| cellanno | Specified metadata name, must exist in rds. Note: cellanno column cannot contain "/" |
TIP
Using the example data, it took about 28 minutes to run in a 4core16GB environment.
Results Interpretation
NOTE
What results are obtained after running? How should they be understood? Next, we will briefly introduce the result contents.
First, check the result directory. You can see that the output results are divided into different folders according to the specified clusters. Taking cluster0 as an example:
tree -L 1 result/cluster0/
result/cluster0/
├── A3SS.MATS.JCEC.txt
├── A5SS.MATS.JCEC.txt
├── demo.b1.txt
├── demo.b2.txt
├── demo_cluster0_A3SS.MATS.JC.txt
├── demo_cluster0_A5SS.MATS.JC.txt
├── demo_cluster0_MXE.MATS.JC.txt
├── demo_cluster0_RI.MATS.JC.txt
├── demo_cluster0_SE.MATS.JC.txt
├── fromGTF.A3SS.txt
├── fromGTF.A5SS.txt
├── fromGTF.MXE.txt
├── fromGTF.novelJunction.A3SS.txt
├── fromGTF.novelJunction.A5SS.txt
├── fromGTF.novelJunction.MXE.txt
├── fromGTF.novelJunction.RI.txt
├── fromGTF.novelJunction.SE.txt
├── fromGTF.novelSpliceSite.A3SS.txt
├── fromGTF.novelSpliceSite.A5SS.txt
├── fromGTF.novelSpliceSite.MXE.txt
├── fromGTF.novelSpliceSite.RI.txt
├── fromGTF.novelSpliceSite.SE.txt
├── fromGTF.RI.txt
├── fromGTF.SE.txt
├── JCEC.raw.input.A3SS.txt
├── JCEC.raw.input.A5SS.txt
├── JCEC.raw.input.MXE.txt
├── JCEC.raw.input.RI.txt
├── JCEC.raw.input.SE.txt
├── JC.raw.input.A3SS.txt
├── JC.raw.input.A5SS.txt
├── JC.raw.input.MXE.txt
├── JC.raw.input.RI.txt
├── JC.raw.input.SE.txt
├── MXE.MATS.JCEC.txt
├── RI.MATS.JCEC.txt
├── SE.MATS.JCEC.txt
├── summary.txt
└── tmp
1 directory, 38 filesThe above txt files are the results of differential alternative splicing analysis of cluster0 relative to other clusters by rMATS. For details about each file, please refer to the rMATS official documentation: rmats-turbo/README.md at v4.3.0 · Xinglab/rmats-turbo (github.com)
Here, we introduce the main output result contents:
I. Main Results demo_cluster0_*.MATS.JC.txt
This result is divided into five files: SE, A5SS, A3SS, MXE, and RI, corresponding to five types of alternative splicing.
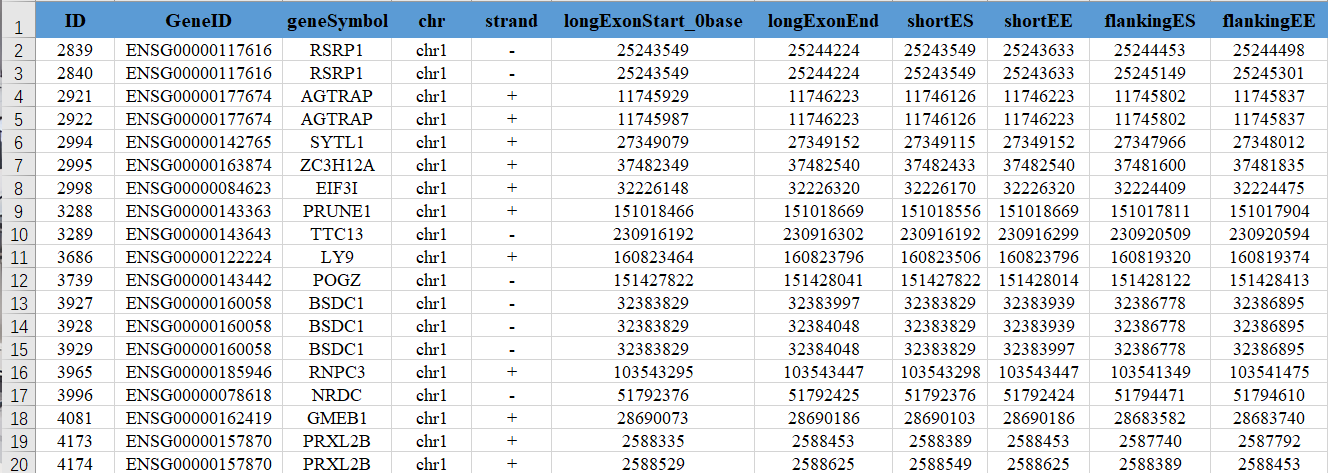
The content includes the start and end positions of each splicing event, detailed information of aligned reads, and calculates the differential levels of each splicing event between cluster0 and all other clusters. The column descriptions are:
(1) ID: rMATS event id
(2) GeneID: Gene ID where the alternative splicing event occurs
(3) geneSymbol: Gene name where the alternative splicing event occurs
(4) chr: Chromosome where the alternative splicing event occurs
(5) strand: Direction of the chromosome strand where the alternative splicing event occurs
(6) ExonStart_0base: Start position of the skipped exon in the alternative splicing event, counting from 0
(7) ExonEnd: End position of the skipped exon in the alternative splicing event
(8) upstreamES: Start position of the upstream exon of the skipped exon in the alternative splicing event
(9) upstreamEE: End position of the upstream exon of the skipped exon in the alternative splicing event
(10) downstreamES: Start position of the downstream exon of the skipped exon in the alternative splicing event
(11) downstreamEE: End position of the downstream exon of the skipped exon in the alternative splicing event
(12) ID: rMATS event id
(13) IJC_SAMPLE_1: Count number of sample one under inclusion junction (IJC), results of repeated samples separated by commas
(14) SJC_SAMPLE_1: Count number of sample one under skipping junction (SJC), results of repeated samples separated by commas
(15) IJC_SAMPLE_2: Count number of sample two under inclusion junction (IJC), results of repeated samples separated by commas
(16) SJC_SAMPLE_2: Count number of sample two under skipping junction (SJC), results of repeated samples separated by commas
(17) IncFormLen: Effective length of Exon Inclusion Isoform in the alternative splicing event
(18) SkipFormLen: Effective length of Exon Skipping Isoform in the alternative splicing event
(19) PValue: P-value of significance of differential expression of alternative splicing events between two groups of samples
(20) FDR: FDR value of significance of differential expression of alternative splicing events
(21) IncLevel1: Ratio of Exon Inclusion Isoform to total expression of two Isoforms in the treatment group
(22) IncLevel2: Ratio of Exon Inclusion Isoform to total expression of two Isoforms in the control group
(23) IncLevelDifference: Difference between IncLevel1 and IncLevel2
II. Main Results summary.txt

This file is a summary of differential alternative splicing events in cluster0, counting the number of alternative splicing events with FDR < 0.05.
Where SignificantEventsJC/JCEC is the sum of SigEventsJC/JCECSample1HigherInclusion and SigEventsJC/JCECSample2HigherInclusion. JC and JCEC refer to results obtained from two different calculation methods respectively.
TIP
For more result queries and understanding, please visit the rMATS official website https://github.com/Xinglab/rmats-turbo/blob/v4.3.0/README.md
References:
Shen S., Park JW., Lu ZX., Lin L., Henry MD., Wu YN., Zhou Q., Xing Y. rMATS: Robust and Flexible Detection of Differential Alternative Splicing from Replicate RNA-Seq Data. PNAS, 111(51):E5593-601. doi: 10.1073/pnas.1419161111