FAST Fusion Module Documentation
Introduction:
NOTE
This document provides instructions for using the Fusion Module for FAST data, describing how to obtain the code and data for the fusion module, along with running examples and result explanations.
Fusion Analysis:
IMPORTANT
Gene fusion refers to structural changes that occur during chromosomal rearrangement, where two or more genes form a new fusion gene. Here, we use STAR-Fusion software for analysis. The software first aligns RNA-seq reads to the reference genome, identifies potential fusion gene pairs, and then generates a high-confidence list of fusion gene candidates through further filtering and validation steps. The junction-reads identified by STAR-Fusion software can be mapped to single cells based on their barcode and UMI tag information, which will serve as fusion gene labels for cells, enabling visualization or other downstream analyses.
I. Data Acquisition:
1) Reference Genome:
IMPORTANT
Running STAR-Fusion requires a pre-built genome. Reference genomes for human and mouse can be obtained below. For other species, please refer to installing star fusion · STAR-Fusion/STAR-Fusion Wiki.
NOTE
Files are large, approximately 30G each. Please download as needed:
# human
wget -c -O Fusion_refdata_human_GRCh38.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_refdata_human_GRCh38.tar.gz"
# Decompress
mkdir Fusion_refdata_human_GRCh38
tar -zxvf Fusion_refdata_human_GRCh38.tar.gz -C Fusion_refdata_human_GRCh38# mouse
wget -c -O Fusion_refdata_mouse_mm10.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_refdata_mouse_mm10.tar.gz"
# Decompress
mkdir Fusion_refdata_mouse_mm10
tar -zxvf Fusion_refdata_mouse_mm10.tar.gz -C Fusion_refdata_mouse_mm102) Environment:
CAUTION
The running environment needs to include seeksoultools, STAR-Fusion, and other required software.
# seeksoultools
wget -c -O seeksoultools_fast_mut_dev.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/seeksoultools_fast_mut_dev.tar.gz"
# Decompress
mkdir seeksoultools
tar -zxvf seeksoultools_fast_mut_dev.tar.gz -C seeksoultools
source seeksoultools/bin/activate
seeksoultools/bin/conda-unpack# Fusion_env
wget -c -O Fusion_env.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_env.tar.gz"
# Decompress
mkdir Fusion_env
tar -zxvf Fusion_env.tar.gz -C Fusion_env
source Fusion_env/bin/activate
Fusion_env/bin/conda-unpack3) Code:
NOTE
Analysis code and test data (human cellline, BCR-ABL1 fusion) can be obtained here:
wget -c -O Fusion_code.tar.gz "https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/Fusion.v1/Fusion_code.tar.gz"
tar -zxvf Fusion_code.tar.gzII. Running Example:
# Add custom tool directories to PATH
# Note: export seeksoultools first
export PATH="`pwd`/seeksoultools/bin:$PATH"
# making Fusion_env highest priority
export PATH="`pwd`/Fusion_env/bin:$PATH"
bash Fusion_code/Fusion.sh \
-s demo \
-l Fusion_code/testdata/BCR_ABL1_R1.fastq.gz \
-r Fusion_code/testdata/BCR_ABL1_R2.fastq.gz \
-c 8 \
-o ./output \
-g Fusion_refdata_human_GRCh38/ctat_genome_lib_build_dir \
-t Fusion_code/testdata/cellline.rdsParameter Description:
| parameter | full-name | description |
|---|---|---|
| s | sample | sample name, required |
| l | R1_fq | raw-fastq data for R1; gz compression format, required |
| r | R2_fq | raw-fastq data for R2; gz compression format, required |
| c | CPU | CPU (default: 8) |
| o | outdir | path for output (default: ./output) |
| g | genome_lib_dir | ref-data for STAR-Fusion, required |
| t | rds | Seurat Object, required; meta.data for this object needs to include the "Sample" column |
Notes:
WARNING
- This module is limited to single sample analysis. For multiple samples, please run scripts separately;
- Input R1/R2 must be FAST/panel rawdata;
- When input with panel data, the rds for the "-t" parameter must be the rds from single-cell quality control of the corresponding FAST data;
- Fusion analysis requires large resources. The larger the sequencing data, the more time and resources required. For 100G of sequencing data, it is recommended to use 32C128GB or more.
III. Running Process
NOTE
After quality control of raw sequencing data using fastp and primer extraction using seeksoultools to obtain clean data, the STAT-Fusion program is called to identify fusion events. Based on the barcode and UMI tag information on JunctionReads, we can identify which cells contain fusion reads and how many UMIs detect fusion reads. These are used as new labels for cells and can be used for downstream analysis. Fusions with JunctionReads greater than or equal to 10 and cell count greater than or equal to 2 are filtered.
IV. Main Results Description
TIP
The following describes the main output results. For more detailed descriptions of output results, please refer to the STAR-Fusion official website Home · STAR-Fusion/STAR-Fusion Wiki.
STAR/sample/star-fusion.fusion_predictions.tsv

IMPORTANT
This table contains all identified fusion gene results, detailing the breakpoints, supporting reads count, left and right region information, and PFAM database information for each fusion. The fusion breakpoint positions can be obtained from LeftBreakpoint and RightBreakpoint, which are also the unique identifiers for subsequent fusion differentiation. The more reads supporting the fusion and satisfy biologically meaningful the splicing pattern, the more reliable the result.
(1) X.FusionName: Fusion gene
(2) JunctionReadCount: Number of reads that can be split and matched to both sides of the fusion gene at the assumed fusion junction
(3) SpanningFragCount: Number of reads containing fusion junctions, where R1 and R2 ends of the reads correspond to different genes
(4) est_J: Original junction read count (estimated value considering multiple alignments and fusion transcript diversity)
(5) est_S: Original spanning read count (estimated value considering multiple alignments and fusion transcript diversity)
(6) SpliceType: Whether the fusion gene breakpoint appears at the reference exon junction provided by reference transcript structure annotation (e.g., gencode)
(7) LeftGene: Left gene of the fusion
(8) LeftBreakpoint: Left chromosome position information of the fusion gene breakpoint (GRCh38)
(9) RightGene: Right gene of the fusion
(10) RightBreakpoint: Right chromosome position information of the fusion gene breakpoint (GRCh38)
(11) LargeAnchorSupport: Whether there are reads with longer base sequences (≥25) matching on both sides of the assumed breakpoint. Fusion genes lacking LargeAnchorSupport are usually false positives
(12) FFPM: Normalized result of reads supporting fusion, i.e., fusion amount per million total reads
(13) LeftBreakDinuc: Two nucleotides adjacent to the left of the breakpoint. If the left/right junction result is GT-AG or GC-AG, it is likely a splicing breakpoint
(14) LeftBreakEntropy: Shannon entropy of 15 exonic bases to the left of the breakpoint (0-2, low entropy sites are usually considered less reliable breakpoints)
(15) RightBreakDinuc: Two nucleotides adjacent to the right of the breakpoint
(16) RightBreakEntropy: Shannon entropy of 15 exonic bases to the right of the breakpoint
(17) annots: Simple annotation of fusion transcriptsoutput/plots/*.png
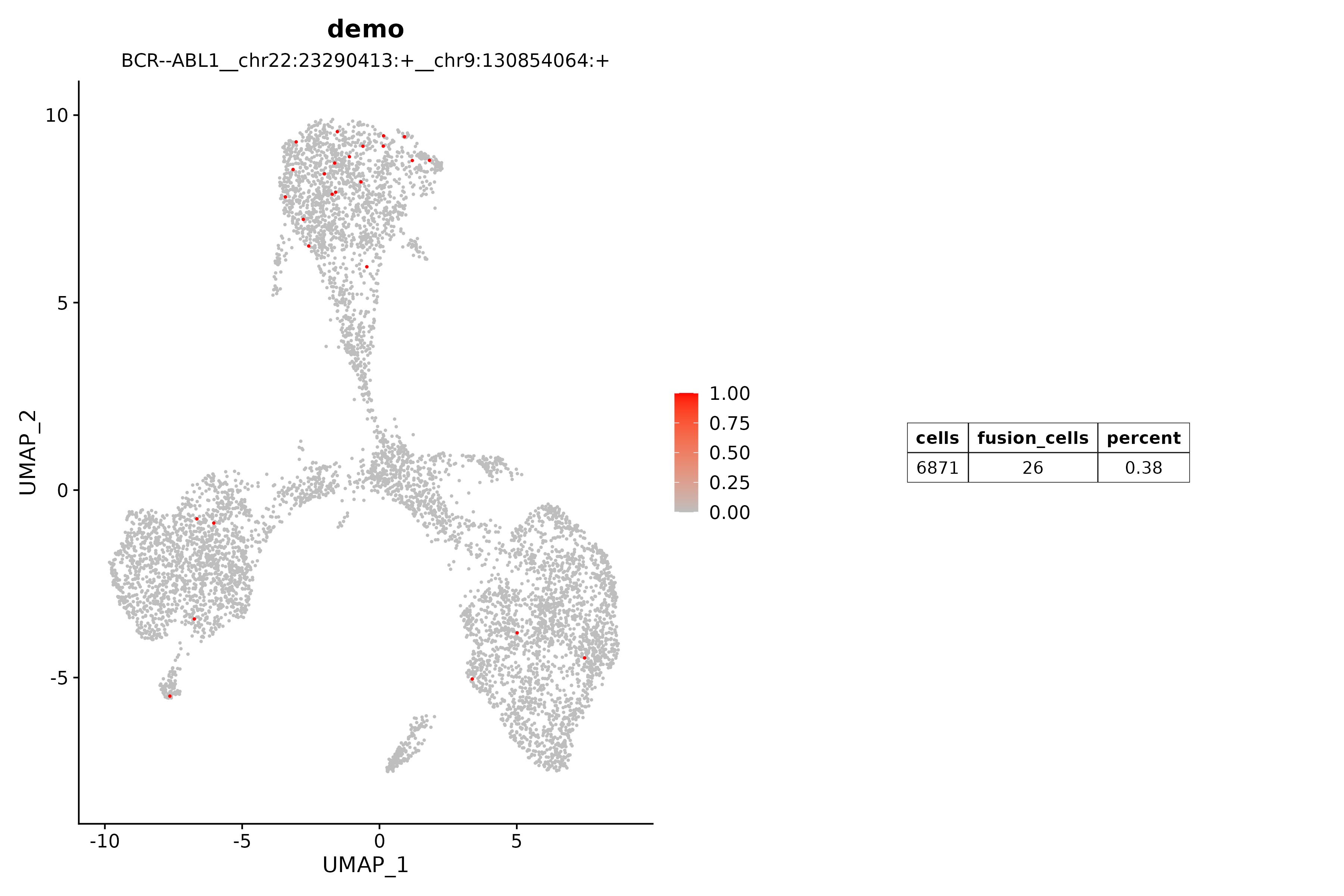
NOTE
For each fusion, umap and tsne plots are displayed. numbers represent the number of UMIs detecting the fusion, and the redder the color, the higher the fusion expression value in that cell. The table on the right shows the proportion of cells with the fusion, where cells represents the total cell count, fusion_cells represents the number of cells with the fusion, and percent represents the percentage.