Cancer Cell Identification Based on CNV and Mutations
Introduction
NOTE
Purpose: To identify cancer cells based on mutation information and CNV results.
Background: In single-cell transcriptome data analysis, conventional methods for identifying cancer cells include copy number variation (CNV) inference, marker gene analysis, and clustering analysis. The paper "Variant calling enhances the identification of cancer cells in single-cell RNA sequencing data" proposes using mutations and CNV information for joint analysis. This approach not only analyzes cancer cell characteristics through mutations and copy number variations but more importantly, can identify cancer cells from cell populations with low copy number variations, thus providing more valuable results.

Cancer Cell Identification Analysis Pipeline
Analysis Approach:
IMPORTANT
- Obtain cell CNV scores from inferCNV analysis;
- Obtain cell mutation matrix from FAST quality control analysis, for mutation counts and annotation information;
- Download cancer gene table from OncoKB database for filtering cancer-related genes;
- Mutation types from FAST quality control analysis include: "frameshift_variant", "inframe_deletion", "inframe_insertion", "missense_variant", "start_lost", "stop_gained", which already meet the requirements;
- Filter pathogenic mutations based on mutation annotation information table;
- Exclude HLA gene-related mutations;
- Obtain mutation counts and CNV results for reference cells and target cells respectively;
- Calculate the distribution position of target cell values in normal cells;
- With specific threshold, target cells meeting either CNV results or mutation counts above threshold are considered cancer cells.
Input Data:
NOTE
- Sample alt mutation matrix, mutation information table (FAST quality control analysis)
- CNV results (CNV_scores.xls table from inferCNV advanced analysis results, must include cells to be analyzed, no downsampling)
- Specify reference cells and target cells
- Specify judgment threshold (default 0.99)
Demo data is provided and can be downloaded and decompressed directly, or obtained by executing commands in the terminal environment:
#run in terminal
wget https://seekgene-public.oss-cn-beijing.aliyuncs.com/software/FAST/mut_and_cnv_demodata.zip
# decompress
unzip mut_and_cnv_demodata.zipRunning Process:
Enter the following code in R environment:
# run in R
#load packages
library(readr)
library(Seurat)
library(dplyr)
library(ggplot2)
# load data
obj = readRDS("mut_and_cnv_demodata/cellline.rds")
mut_matrix = read.delim("mut_and_cnv_demodata/cellline.snp_indel.alt_UMI.matrix",
header = TRUE,row.names = 1)
mut_info = read.delim("mut_and_cnv_demodata/cellline.anno.cluster.xls",header = TRUE)
CNV_score = read.csv("mut_and_cnv_demodata/cellline_CNV_score.csv")
#specific ref cell and target cell
ref_cell=colnames(obj)[obj$celltype == "ref_cell"]
target_cell=colnames(obj)[obj$celltype == "target_cell"]
#load R code
source("mut_and_cnv_demodata/calling_cancer.R")TIP
Run commands to obtain results:
res=calling_cancer(mut_matrix = mut_matrix,
mut_info = mut_info,
cnv_info = CNV_score,
ref_cells = ref_cell,
target_cells = target_cell,
threshold = 0.99)
p1=callling_cancer_plot(res)
cancer_cell=res$cancer_cell
cancer_label=rep("other",ncol(obj))
cancer_label[match(cancer_cell,colnames(obj))]="cancer_cell"
obj=AddMetaData(obj,metadata=cancer_label,col.name="cancer_label")
p2=DimPlot(obj,group.by="cancer_label",label=T)Analysis Results:
p1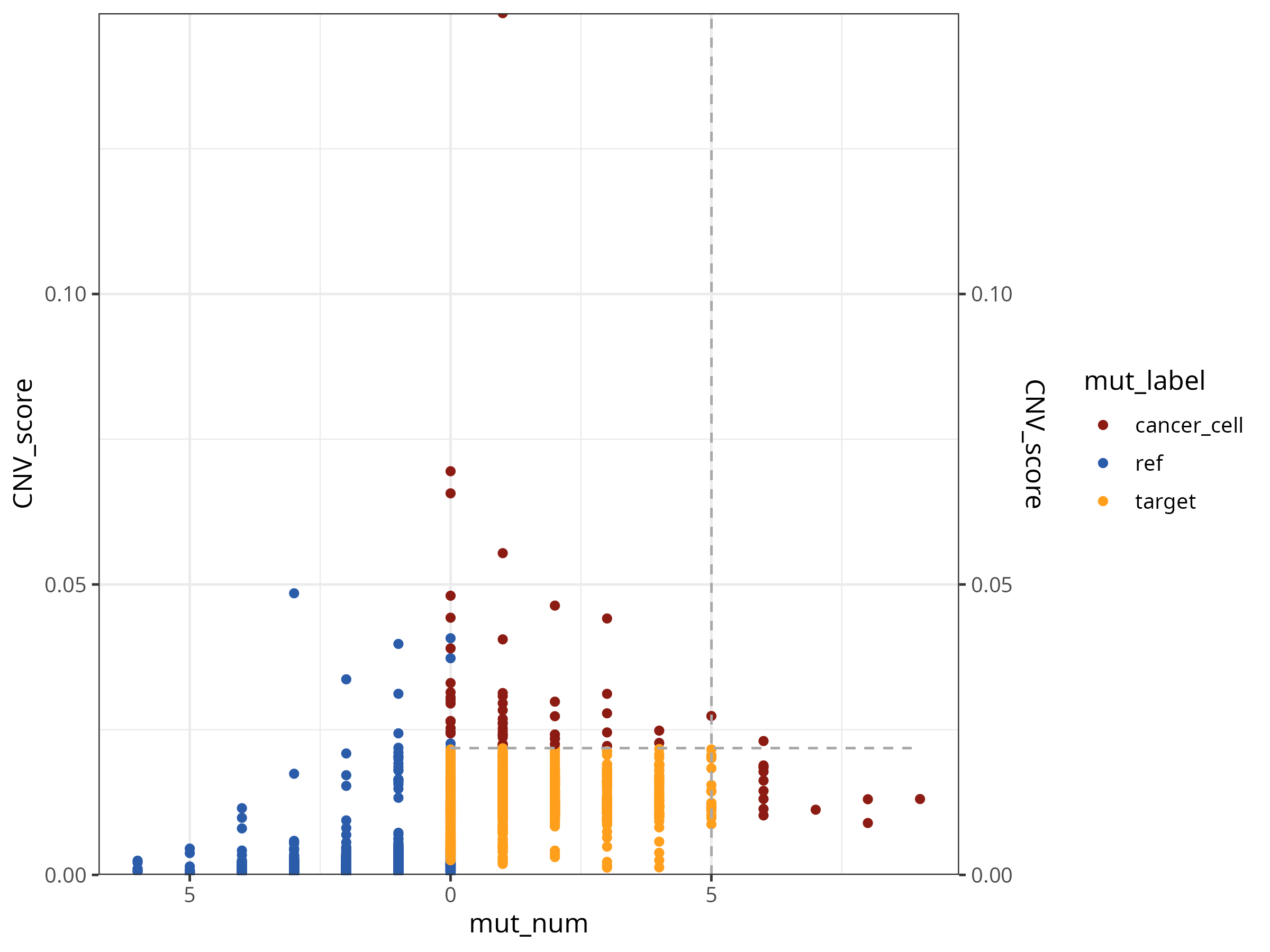
Shows mutation and CNV information for reference cells and target cells, with labeled cancer cells. The dashed lines show the judgment thresholds for mutations and CNV respectively.
p2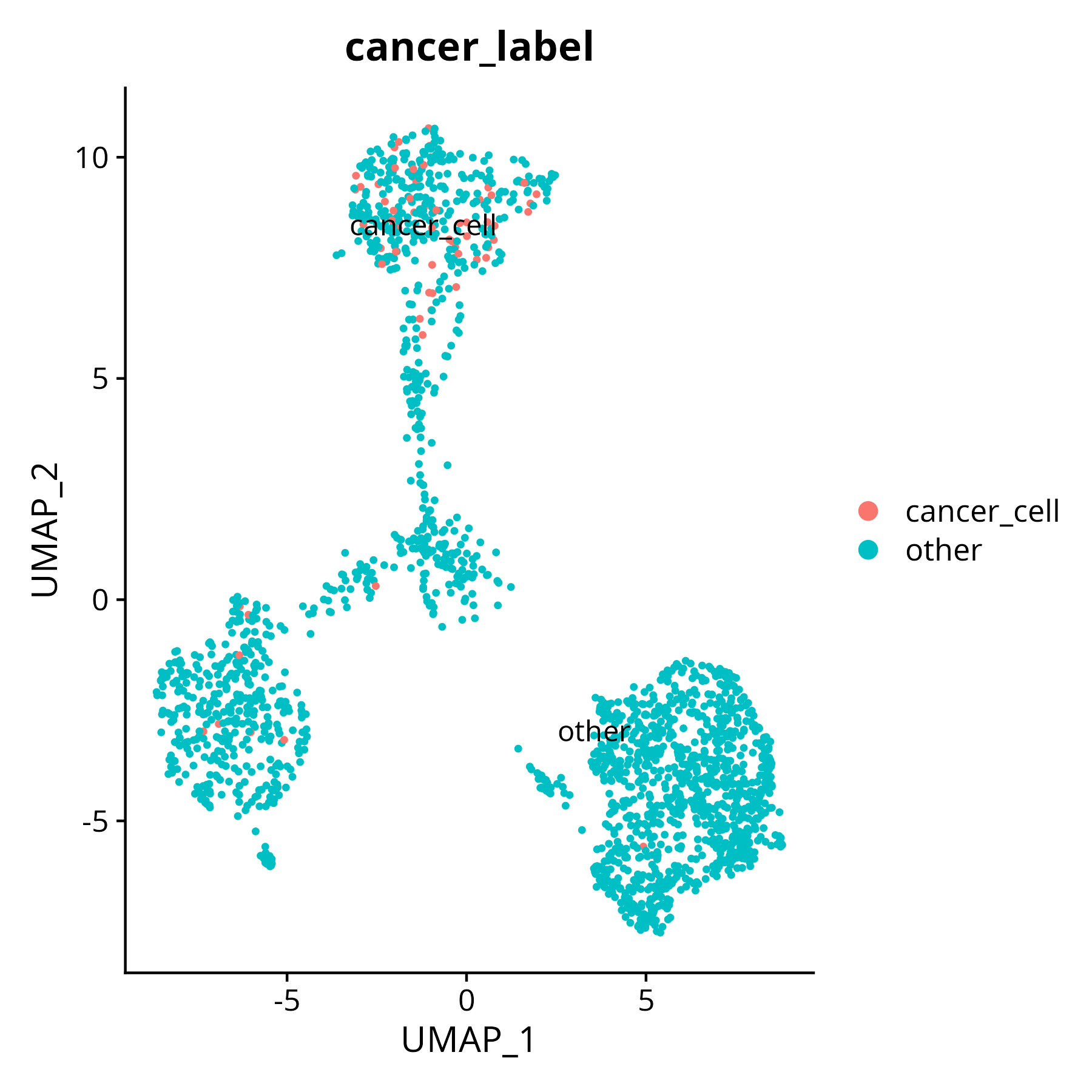
Shows the addition of identified cancer cells as new labels to the object, which can be used for downstream analysis.
Save images:
ggsave(p1, file = "cellline_calling_cancer.png", width = 8, height = 6)
ggsave(p2, file = "cellline_cancer_label.png", width = 6, height = 6)References:
Gasper W, Rossi F, Ligorio M, Ghersi D (2022) Variant calling enhances the identification of cancer cells in single-cell RNA sequencing data. PLOS Computational Biology 18(10): e1010576. https://doi.org/10.1371/journal.pcbi.1010576
High throughput detection of variation in single-cell whole transcriptome through streamlined scFAST-seq, Guoqin Sang, Jiaxin Chen, Meng Zhao, Huanhuan Shi, Jinhuan Han, Jiacheng Sun, Ying Guan, Xingyong Ma, Guangxin Zhang, Yuyan Gong, Yi Zhao, Shaozhuo Jiao, bioRxiv 2023.03.19.533382; doi: https://doi.org/10.1101/2023.03.19.533382
R Package Versions:
NOTE
R 4.4.1
readr 2.1.5
Seurat 5.1.0
dplyr 1.1.4
ggplot2 3.5.1